GC-IMS vs. Lab-Based Methods: A New Paradigm for Rapid On-Site Drug and VOC Screening
This article explores the transformative potential of Gas Chromatography-Ion Mobility Spectrometry (GC-IMS) for rapid on-site analysis, directly comparing it with established laboratory-based methods like GC-MS.
GC-IMS vs. Lab-Based Methods: A New Paradigm for Rapid On-Site Drug and VOC Screening
Abstract
This article explores the transformative potential of Gas Chromatography-Ion Mobility Spectrometry (GC-IMS) for rapid on-site analysis, directly comparing it with established laboratory-based methods like GC-MS. Tailored for researchers and drug development professionals, we dissect the foundational principles of GC-IMS, its operational methodologies in diverse applications—from clandestine drug laboratory detection to food quality control—and critical troubleshooting strategies for field deployment. A rigorous validation and comparative analysis provides a clear framework for selecting the appropriate technology based on sensitivity, speed, and operational requirements. The synthesis concludes that GC-IMS is not a mere replacement but a powerful complementary tool, with profound implications for accelerating forensic investigations, clinical diagnostics, and quality control processes where speed and portability are paramount.
Understanding GC-IMS: Core Principles and Advantages for Field Deployment
Gas Chromatography-Ion Mobility Spectrometry (GC-IMS) represents a powerful hybrid analytical technique that couples the separation capabilities of gas chromatography with the sensitive detection properties of ion mobility spectrometry. This combination creates a synergistic system particularly suited for the rapid, sensitive analysis of volatile organic compounds (VOCs). The operational duo of GC and IMS provides a compelling solution for applications ranging from food quality assessment to medical diagnostics, offering distinct advantages for rapid on-site screening scenarios where traditional laboratory methods may be too slow, expensive, or non-portable [1] [2].
The fundamental strength of this hybrid system lies in the complementary nature of its components. The GC first separates complex mixtures into their individual chemical constituents based on their partitioning behavior between a mobile gas phase and a stationary phase, a process that provides high resolution for volatile compounds. The separated analytes then undergo ionization and a second separation in the IMS drift tube based on their size, charge, and shape as they move through a buffer gas under an electric field [3] [4]. This two-dimensional separation delivers enhanced specificity compared to either technique alone, effectively reducing false positives and providing a more comprehensive chemical fingerprint of samples, all while maintaining the potential for portability and rapid analysis that is crucial for on-site applications [4] [2].
Operational Principles of the GC-IMS Duo
Gas Chromatography: The First Dimension of Separation
The gas chromatograph serves as the first dimension of separation in the GC-IMS system. Its primary function is to separate complex volatile mixtures into their individual components before they enter the detector. The separation process occurs within a chromatographic column, where a carrier gas (such as helium or nitrogen) transports the vaporized sample. As the sample moves through the column, its components interact with the stationary phase lining the column interior, causing each compound to elute at a characteristic retention time based on its chemical properties and affinity for the stationary phase [5]. This process effectively spreads the sample components over time, transforming a complex mixture into a series of discrete chemical bands emerging from the column.
The efficiency of this chromatographic separation is critical for the overall performance of the GC-IMS system. Proper GC separation ensures that the number of compounds entering the IMS detector simultaneously is minimized, which is crucial because IMS detection is susceptible to matrix effects and competitive ionization when multiple analytes are present together [4]. Without effective GC pre-separation, ionization suppression can occur, where certain compounds dominate the ionization process and suppress the signals of other, potentially important, analytes. The GC column thereby acts as a temporal filter, delivering simplified chemical packets to the IMS for subsequent detection and mobility-based separation [3].
Ion Mobility Spectrometry: The Second Dimension of Detection
Following GC separation, the eluting compounds enter the ion mobility spectrometer, which provides the second dimension of analysis. The IMS process begins in the ionization region, where a radioactive source (typically tritium or nickel-63) generates reactant ions from the carrier gas. These reactant ions, often clusters of H3O+(H2O)n, subsequently ionize the analyte molecules through charge-transfer reactions, forming protonated monomer or dimer ions [3] [4]. The newly formed analyte ions are then introduced into the drift tube via an ion shutter or gate.
Inside the drift tube, a constant electric field accelerates the ions through a counter-flowing drift gas (typically nitrogen or clean air). During their journey, the ions experience frequent collisions with the drift gas molecules. The resulting drift velocity achieved by each ion depends on its collision cross-section, mass, and charge. Smaller, more compact ions experience fewer collisions and reach the detector faster than larger, bulkier ions. The detector records the arrival times of the ions, producing a mobility spectrum where each peak corresponds to a specific ion species [3] [4]. This separation based on ion mobility in the gas phase occurs remarkably quickly, typically within tens of milliseconds, allowing multiple mobility spectra to be acquired across a single GC peak [4].
Creating a Two-Dimensional Analytical Fingerprint
The powerful synergy of GC-IMS emerges from the orthogonality of its two separation dimensions. The system generates a three-dimensional data output where the x-axis represents the GC retention time, the y-axis represents the IMS drift time, and the signal intensity is represented by a color gradient or contour plot [2]. This creates a comprehensive two-dimensional fingerprint of the sample's volatile composition, with each compound represented by a spot at specific retention and drift time coordinates.
This two-dimensional separation provides significantly higher peak capacity than either technique alone, dramatically improving the ability to resolve complex mixtures. The technique is particularly effective for distinguishing isomeric compounds that may co-elute in GC but have different collision cross-sections detectable by IMS [2]. Furthermore, the high repetition rate of IMS measurement (typically 10-100 spectra per second) ensures that even narrow GC peaks are adequately sampled across both dimensions, preserving the chromatographic resolution while adding the mobility dimension for enhanced compound identification and characterization [4].
Comparative Performance: GC-IMS vs. GC-MS and Other Alternatives
When evaluating analytical techniques for specific applications, understanding relative performance characteristics is essential. The following comparison highlights how GC-IMS positions itself against the established laboratory gold standard, GC-MS, and other alternatives.
Table 1: Technical Comparison of GC-IMS versus GC-MS
| Performance Parameter | GC-IMS | GC-MS |
|---|---|---|
| Detection Sensitivity | Approx. 10× more sensitive for certain compounds (picogram/tube range) [6] | Nanogram/tube range [6] |
| Linear Dynamic Range | 1-2 orders of magnitude (after linearization) [6] | 3 orders of magnitude (up to 1000 ng/tube) [6] |
| Analysis Speed | Rapid (seconds to minutes for IMS detection) [1] | Slower (typically minutes to hours) [2] |
| Portability | Portable systems available; no vacuum required [3] [1] | Typically benchtop; requires vacuum system [3] |
| Operational Costs | Lower maintenance; minimal consumables [1] [4] | Higher maintenance; vacuum pumps, etc. [3] |
| Sample Throughput | High (fast IMS cycles enable rapid screening) [2] | Moderate to low (longer analysis times) [2] |
| Identification Capability | Limited databases; often requires reference standards [6] | Extensive spectral libraries available [3] [6] |
| Ease of Operation | Minimal sample preparation; user-friendly [1] [2] | Often requires complex sample preparation [2] |
GC-IMS demonstrates distinct advantages in applications requiring rapid screening, high sensitivity, and portability. The technology's superior sensitivity, often approximately ten times greater than MS for certain compounds, makes it particularly suitable for trace-level analysis [6]. Furthermore, the elimination of vacuum systems required by MS instruments enables the development of portable GC-IMS systems that can be deployed for on-site analysis, a crucial advantage for field applications [3]. The technique's speed, with IMS separation occurring in milliseconds, facilitates high-throughput screening scenarios where time is critical [1] [2].
However, GC-MS maintains important advantages in other areas. MS provides a broader linear dynamic range – typically three orders of magnitude compared to one to two for IMS – making it more suitable for quantitative analysis across concentration ranges [6]. Most significantly, GC-MS benefits from extensive, well-established mass spectral libraries that facilitate compound identification, whereas IMS lacks universally available reference databases, often requiring analytical standards for definitive identification [6]. This makes GC-MS preferable for untargeted analysis where comprehensive compound identification is necessary.
Table 2: Method Characteristics Comparison for Different Analytical Scenarios
| Application Scenario | Recommended Technique | Key Rationale |
|---|---|---|
| Rapid On-Site Screening | GC-IMS | Portability, speed, sensitivity for targeted compounds [1] [2] |
| Untargeted Discovery | GC-MS | Comprehensive spectral libraries for unknown identification [3] [6] |
| Quantitative Analysis | GC-MS | Broader linear range, established quantification protocols [6] |
| Isomer Differentiation | GC-IMS | Orthogonal separation based on collision cross-section [2] |
| Field Deployment | GC-IMS | No vacuum requirements, portable configurations available [3] [1] |
Experimental Protocols and Methodologies
Standard GC-IMS Analysis Workflow
A typical GC-IMS analytical protocol involves several standardized steps to ensure reproducibility and data quality. For headspace analysis (HS-GC-IMS), which is commonly used for volatile compound profiling, the methodology begins with sample preparation – often involving homogenization and portioning into headspace vials. The samples are then incubated at a controlled temperature to allow volatile compounds to partition into the headspace. A defined volume of headspace gas is automatically injected into the GC inlet, where it is transported by the carrier gas onto the chromatographic column [2].
The chromatographic separation typically employs capillary columns (conventional diameter or multi-capillary columns for increased sample capacity) with temperature programming to optimize separation efficiency. After GC separation, the eluting compounds enter the IMS drift tube maintained at a constant temperature. The IMS detection parameters – including drift tube temperature, drift gas flow rate, and electric field strength – are optimized for the application. The resulting data is visualized as a 2D fingerprint, with each volatile compound appearing as a spot characterized by its retention time (x-axis) and drift time (y-axis) [3] [2].
Data Processing and Feature Extraction
GC-IMS generates highly dimensional data that requires specialized processing to extract meaningful information. The raw data consists of a three-dimensional structure (retention time, drift time, intensity) for each sample. Data pre-processing typically includes noise reduction, baseline correction, and alignment in both retention and drift time dimensions to correct for run-to-run variations [3]. Following pre-processing, feature extraction is performed to identify and quantify the relevant signals.
Several approaches exist for feature extraction from GC-IMS data. These include: (1) extracting features from the total area of the reactant ion peak chromatogram (RIC); (2) using the full RIC response; (3) analyzing the unfolded sample matrix; or (4) focusing on specific ion peak volumes [3]. The choice of strategy represents a trade-off between the amount of chemical information preserved and the computational effort required. Following feature extraction, multivariate statistical analysis – such as principal component analysis (PCA) or partial least squares-discriminant analysis (PLS-DA) – is often applied to identify patterns and discriminate between sample classes based on their volatile profiles [3].
GC-IMS Analytical Workflow
Application Performance in Research Settings
Food Quality and Authenticity Control
GC-IMS has demonstrated exceptional performance in food quality assessment and authenticity verification. In one notable application, researchers successfully distinguished different quality classes of Iberian ham based on the feeding regime of the pigs (acorn-fed vs. feed-fed) – a differentiation with significant economic implications given the price ratio between categories can exceed one order of magnitude [3]. The GC-IMS analysis revealed distinct volatile profiles that enabled classification with high accuracy, providing a rapid method to combat alimentary fraud in the ham industry [3]. Similar approaches have been applied to olive oil authenticity, honey origin verification, and detection of fish and egg freshness, consistently demonstrating the technique's capability to identify characteristic volatile fingerprints associated with product quality and authenticity [3] [2].
The technology's speed and minimal sample preparation make it particularly suitable for quality control in industrial settings. For instance, studies have monitored flavor changes in agri-food products during storage and processing, and detected microbial contamination before visible spoilage occurs [2]. Compared to traditional GC-MS methods, GC-IMS offers the advantage of rapid analysis cycles and the potential for on-site deployment at production facilities, enabling real-time decision making rather than waiting for laboratory results [2].
Medical Diagnostics and Biomarker Discovery
In medical applications, GC-IMS shows transformative potential for rapid, non-invasive disease diagnostics. The technology has been applied to various human metabolites, including urine, feces, bile, serum, and exhaled breath, for differential detection of diseases and pathogenic microbes [1]. The high sensitivity of IMS detection enables identification of trace-level volatile biomarkers that may be present in biological samples, while the GC pre-separation helps manage the complex matrix effects inherent in such samples [1].
A significant advantage in medical applications is the technique's ability to provide rapid results with minimal sample preparation – crucial for clinical settings. For example, in exhaled breath analysis, GC-IMS can detect biomarkers such as ethanol, isoprene, and acetone, potentially enabling non-invasive screening for various metabolic disorders and respiratory diseases [6]. While GC-MS remains the gold standard for definitive compound identification in research settings, GC-IMS offers a compelling alternative for rapid screening applications where speed and portability may outweigh the need for comprehensive compound identification [1].
Essential Research Reagents and Materials
Successful GC-IMS analysis requires specific reagents and materials optimized for the technique's operational requirements. The following table details key components of the GC-IMS research toolkit.
Table 3: Essential Research Reagents and Materials for GC-IMS
| Item | Function | Technical Specifications |
|---|---|---|
| Drift Gases | Provides inert environment for ion separation | High-purity nitrogen or clean, dry air; requires moisture trap [4] |
| Carrier Gases | Transports sample through GC column | Helium or nitrogen; dry, oxygen-free, inert [5] |
| Chemical Standards | System calibration and compound identification | Ketones, aldehydes, alcohols for calibration; purity ≥95% [6] |
| Derivatization Reagents | Enhances volatility of polar compounds | MSTFA, TMCS; for analyzing highly polar substances [5] |
| Thermal Desorption Tubes | Sample collection and concentration | Tubes with adsorbent materials (Tenax, Carbograph) [6] |
| Reference Compounds | Creating identification databases | Target analytes dissolved in appropriate solvents (e.g., methanol) [6] |
The selection of appropriate drift and carrier gases is critical for maintaining stable operation and achieving optimal separation performance. These gases must be of high purity to prevent contamination of the ionization region and drift tube, which could lead to increased noise and reduced sensitivity. For quantitative work, chemical standards are essential for calibrating both the retention time (GC) and reduced mobility (IMS) scales, enabling reproducible compound identification across different instruments and analysis sessions [6]. The availability of well-characterized standards is particularly important given the current lack of universal IMS databases comparable to those available for GC-MS.
Critical Considerations for Implementation
Analytical Strengths and Advantages
GC-IMS offers several compelling advantages that make it suitable for specific analytical scenarios. Its exceptional sensitivity, with detection limits in the picogram per tube range for certain compounds, enables trace-level analysis of volatile organic compounds [6]. This high sensitivity, combined with the portability of IMS technology (which doesn't require vacuum systems), makes GC-IMS uniquely positioned for on-site applications where laboratory-based instruments cannot be deployed [3] [1]. The technique's speed is another significant advantage, with analysis times typically ranging from seconds to minutes, considerably faster than many conventional GC-MS methods [1] [2].
The orthogonal separation provided by the GC-IMS combination significantly enhances specificity compared to either technique alone. This two-dimensional approach reduces the likelihood of false positives from co-eluting compounds and provides more confident compound identification [4] [2]. Additionally, GC-IMS operation is generally more cost-effective than GC-MS, with lower maintenance requirements and minimal consumables beyond high-purity gases [1] [4]. The technique also requires relatively simple sample preparation compared to many analytical methods, further reducing analysis time and complexity [2].
Limitations and Methodological Constraints
Despite its advantages, GC-IMS does have important limitations that must be considered when selecting an analytical approach. The technique's limited linear dynamic range – typically one to two orders of magnitude even after linearization strategies – can be restrictive for quantitative applications requiring analysis across wide concentration ranges [6]. This contrasts with GC-MS, which often maintains linearity over three orders of magnitude [6]. Perhaps the most significant limitation is the lack of universal reference databases for IMS spectra, necessitating the analysis of authentic standards for definitive compound identification [6]. This makes the technique less suitable for completely untargeted analysis of unknown compounds.
GC-IMS is also susceptible to matrix effects in the ionization region, where competitive ionization can occur when multiple analytes are present simultaneously, potentially suppressing the signals of less-responsive compounds [4]. While the GC pre-separation mitigates this issue, complete elimination is challenging with complex samples. The technique is primarily limited to volatile and semi-volatile compounds, though this is a constraint shared with conventional GC methods. For non-volatile or highly polar compounds, derivatization is often required to increase volatility, adding complexity to the sample preparation process [5].
Analytical Technique Positioning
GC-IMS represents a powerful hybrid analytical tool that successfully merges the separation capabilities of gas chromatography with the sensitive, rapid detection properties of ion mobility spectrometry. This operational duo creates a synergistic system that delivers enhanced specificity through orthogonal separation mechanisms while maintaining the potential for portable deployment – a combination that positions GC-IMS uniquely for rapid on-site screening applications. The technology demonstrates clear advantages in scenarios requiring high sensitivity, rapid analysis, and field deployability, particularly for targeted compound analysis where reference standards are available.
While GC-MS remains the gold standard for comprehensive compound identification and quantitative analysis across broad concentration ranges, GC-IMS establishes its own significant niche in the analytical landscape. As the technology continues to evolve, addressing current limitations around compound identification through expanded databases and standardized protocols will further enhance its utility. For researchers and application scientists requiring rapid, sensitive volatile compound analysis with the flexibility of on-site deployment, GC-IMS offers a compelling analytical solution that effectively bridges the gap between laboratory-based precision and field-based practicality.
Ion Mobility Spectrometry (IMS) has emerged as a powerful analytical technique characterized by exceptional sensitivity, capable of detecting trace-level compounds down to the parts-per-trillion (ppt) range. This inherent sensitivity stems from two core principles: highly efficient chemical ionization processes that gently and effectively ionize a wide range of analytes, and drift time separation that provides an orthogonal dimension to reduce chemical noise. Within the context of rapid on-site screening, Gas Chromatography coupled with IMS (GC-IMS) presents a compelling alternative to traditional lab-based methods like GC-MS, offering a unique balance of high sensitivity, portability, and speed. This guide objectively compares the performance of GC-IMS against other analytical techniques, supported by experimental data and detailed methodologies, to illuminate its specific advantages for field deployment.
The remarkable sensitivity of IMS is not the result of a single feature, but rather the synergistic effect of its fundamental operating principles. At its core, IMS separates and identifies ionized molecules in the gas phase based on their mobility in a carrier buffer gas under the influence of an electric field [7]. The sensitivity originates from an efficient ionization mechanism that maximizes the conversion of neutral analyte molecules into ions, followed by a time-based separation that effectively distinguishes these analyte ions from background chemical noise.
The fundamental relationship defining an ion's behavior in IMS is its drift velocity ((vd)), which is proportional to the electric field strength ((E)) via its ion mobility ((K)) [8] [7]: [ vd = K E ] This mobility, (K), is a unique characteristic for each ion-gas pair and is inversely related to the ion's collision cross section (CCS, or Ω), a measure of its three-dimensional size and shape in the gas phase [8]. The ability to separate ions based on this distinct physical property, on a millisecond timescale, provides IMS with its high orthogonality and low-limit of detection.
The Chemistry of Ionization: A Mechanism Designed for Efficiency
The initial and most critical step dictating IMS sensitivity is the ionization of analyte molecules. Unlike high-energy electron impact ionization used in some mass spectrometers, which can cause extensive fragmentation, IMS typically employs softer chemical ionization (CI). This process is designed to transfer charge gently, preserving the molecular ion and thereby increasing the signal for the target compound.
Ionization Mechanism and Reactant Ions
In positive mode operation, which is common for detecting volatile organic compounds (VOCs), the process begins with the creation of reactant ions [9]:
- Primary Ion Formation: A ionization source (e.g., a small radioactive β-emitter like Tritium ((^3)H) or Nickel-63 ((^{63})Ni)) emits electrons that interact with the drift gas (often Nitrogen or purified air), generating primary ions [9] [6].
- Reactant Ion Formation: These primary ions rapidly undergo a series of reactions with trace water vapor and other molecules in the drift tube, forming stable reactant ions, such as protonated water clusters ((H2O)nH^+) [9]. These clusters appear as an intense peak in the spectrum known as the Reactant Ion Peak (RIP), which acts as a reagent and baseline.
- Analyte Ionization: When analyte molecules (M) enter the reaction region, they interact with the reactant ions via proton transfer or cluster formation reactions [9]:
- Protonated Monomer Formation: ( M + (H2O)nH^+ \leftrightarrow M(H2O){n-x}H^+ + x H_2O )
- Protonated Dimer Formation (at higher analyte concentrations): ( M + M(H2O){n-x}H^+ \leftrightarrow M2(H2O){n-(x+i)}H^+ + i H2O )
This CI process is extremely efficient, leading to the excellent sensitivity IMS is known for [4]. The use of dopants can further enhance selectivity and sensitivity for specific target compounds, such as chemical warfare agents or explosives, by promoting their ionization over background interferents [7].
Diagram: Ionization Process and Ion Separation in a Drift Tube IMS
Drift Time Separation: Isolating Signal from Noise
Following ionization, the separation of ions in the drift tube provides the second critical component of IMS sensitivity. In a classic Drift Tube IMS (DTIMS), a uniform electric field is applied across a tube filled with an inert buffer gas [8] [7].
- Separation Principle: Ions are injected into the tube as a narrow pulse and are propelled by the electric field. Their velocity is determined by their mobility (K), which depends on their mass, charge, and most importantly, their collision cross section (CCS) [8]. Smaller, more compact ions experience fewer collisions with the buffer gas and traverse the tube faster than larger, bulkier ions.
- Drift Time Measurement: The time taken for an ion to travel the length of the drift tube is its drift time ((tD)). The ion mobility (K) can be calculated directly from this measurement using the equation [7]: [ K = \frac{L^2}{tD U} ] where (L) is the drift length and (U) is the total voltage drop.
- Enhancing Signal-to-Noise: This time-dispersive separation is crucial for sensitivity. It spatially and temporally separates analyte ions from the intense RIP and other chemical background ions. This orthogonal separation dimension reduces spectral interference, allowing the signal from a low-concentration analyte to be distinguished from the chemical noise, thereby lowering the practical limit of detection [10].
Experimental Protocols: Validating IMS Performance
To quantitatively assess the sensitivity and performance of IMS, standardized experimental protocols are essential. The following methodology, derived from recent literature, outlines a robust approach for evaluating GC-IMS.
Protocol: Long-Term Stability and Sensitivity Assessment of GC-IMS
Objective: To determine the long-term reproducibility, signal stability, and limits of detection for a GC-IMS system using ketone standards [6].
Materials and Reagents:
- Standard Solutions: Ketone mix (e.g., 2-butanone, 2-pentanone, 2-hexanone, 2-heptanone, 2-octanone) in methanol [6].
- Thermal Desorption (TD) Tubes: Containing adsorbent material such as Tenax TA.
- GC-IMS Instrument: Equipped with a Tritium or (^{63})Ni ionization source and a defined drift tube.
- GC Column: FS-SE-54-CB-1 or equivalent.
- Drift Gas: High-purity Nitrogen or dried, purified air.
Methodology:
- Sample Introduction: Liquid standards are spiked onto TD tubes using a mobile, temperature- and flow-controlled sampling unit to ensure highly reproducible adsorption [6].
- Thermal Desorption & Separation: TD tubes are heated to release analytes into the carrier gas stream. Compounds are pre-separated by the GC column based on their partitioning between the mobile and stationary phases.
- Ionization and Drift Time Separation: Eluting compounds enter the IMS, are ionized via the CI process, and are separated in the drift tube based on their mobility.
- Data Acquisition: Spectra are collected continuously. Each analyte is identified by its GC retention time ((rt)) and IMS drift time ((dt)), creating a 2D fingerprint ((rt), (dt)) [9].
- Long-Term Study: Repeated measurements are conducted over an extended period (e.g., 16 months) to assess signal intensity, retention time, and drift time stability [6].
Key Performance Metrics:
- Limit of Detection (LOD): The lowest analyte concentration that can be reliably detected.
- Signal Stability: Relative Standard Deviation (RSD%) of normalized signal intensities over time.
- Reproducibility: RSD% of GC retention times and IMS drift times.
Comparative Performance Data: IMS vs. MS
Direct comparative studies provide the most objective evidence for the performance characteristics of different analytical techniques. The following data summarizes findings from a systematic evaluation of a TD-GC-MS-IMS system.
Table 1: Quantitative Performance Comparison of IMS and MS Detectors in VOC Analysis [6]
| Performance Parameter | Ion Mobility Spectrometry (IMS) | Mass Spectrometry (MS) |
|---|---|---|
| Typical Limit of Detection (LOD) | Low picogram per tube range (Approx. 10x more sensitive than MS for target VOCs) | ~10x higher than IMS for target VOCs |
| Linear Dynamic Range | 1 to 2 orders of magnitude (e.g., 0.1 to 10 ng/tube) after linearization | >3 orders of magnitude (up to 1000 ng/tube) |
| Long-Term Signal Intensity RSD | 3% to 13% (over 16 months) | 3.0% to 7.6% |
| Retention Time Reproducibility | 0.10% to 0.22% RSD | Comparable to IMS |
| Drift Time Reproducibility | 0.49% to 0.51% RSD | Not Applicable |
| Key Strength | Ultra-high sensitivity, speed, portability | Wide linear range, extensive library identification |
Table 2: Application-Based Comparison: GC-IMS vs. GC-MS for On-Site vs. Laboratory Analysis
| Feature | GC-IMS (On-Site Screening) | GC-MS (Lab-Based Analysis) |
|---|---|---|
| Analysis Speed | Seconds to minutes [11] [7] | Minutes to hours |
| Portability | High (palm-portable systems available) [7] | Low (typically benchtop) |
| Sensitivity | Parts-per-trillion (ppt) to parts-per-billion (ppb) [12] [9] | Parts-per-trillion (ppt) |
| Selectivity & Identification | Good (2D separation: RT & Drift Time) | Excellent (library matching) |
| Operational Cost & Consumables | Lower (drift gas only) [4] | Higher (high-purity gases, maintenance) |
| Ideal Use Case | Rapid screening, hazardous material detection, process monitoring [12] [7] [9] | Definitive identification, complex unknown analysis, quantification over wide range [6] |
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of GC-IMS methods relies on a set of key consumables and reagents.
Table 3: Essential Research Reagent Solutions for GC-IMS Experiments
| Item | Function / Purpose | Example Specifications |
|---|---|---|
| Drift Gas | Inert buffer gas for the drift tube; defines separation environment. | High-purity Nitrogen ((N_2)) or dried, purified air [13] [4]. |
| Chemical Standards | Instrument calibration, quantification, and method development. | Purity ≥95%; e.g., Ketone, Aldehyde, Alcohol mixes in methanol [6]. |
| Thermal Desorption Tubes | Sample collection, concentration, and introduction. | Tubes packed with adsorbent material (e.g., Tenax TA) [6]. |
| Dopant Gases | Enhance selectivity and sensitivity for specific compound classes. | Acetone (for CWAs), Chlorinated solvents (for explosives) [7]. |
| Calibration Kit | For determining reduced ion mobility ((K_0)) and Collision Cross Section (CCS). | A set of compounds with well-characterized mobility values (e.g., ketones) [8]. |
The inherent sensitivity of IMS is a direct consequence of its efficient chemical ionization process and its orthogonal drift time separation. The data confirms that IMS, particularly when coupled with GC, offers a unique combination of ultra-high sensitivity, rapid analysis, and portability that is exceptionally well-suited for rapid on-site screening applications. While laboratory-based GC-MS remains the gold standard for unambiguous identification and quantification over a wide dynamic range, GC-IMS stands out as a powerful alternative for scenarios demanding immediate, sensitive, and on-location results. The choice between these techniques ultimately depends on the specific analytical requirements, balancing the need for speed and sensitivity in the field against the need for definitive identification and broad quantification in the laboratory.
Gas Chromatography coupled with Ion Mobility Spectrometry (GC-IMS) is emerging as a powerful analytical technique that addresses critical limitations of traditional laboratory systems, particularly for rapid on-site screening applications. When compared to established methods like Gas Chromatography-Mass Spectrometry (GC-MS), GC-IMS demonstrates superior performance in portability, analytical speed, and operational cost-effectiveness, while maintaining high sensitivity and reliability. This guide provides an objective comparison of GC-IMS against traditional lab-based systems, supported by experimental data and structured analysis of performance metrics across key differentiation parameters.
Technical Comparison: GC-IMS vs. GC-MS
The following table summarizes the core differentiators between GC-IMS and traditional GC-MS systems based on current research and application data.
Table 1: Key Differentiators Between GC-IMS and GC-MS Systems
| Parameter | GC-IMS | GC-MS (Traditional Lab System) |
|---|---|---|
| Portability | High – Commercially available as portable, miniaturized, and benchtop systems; some designs are chip-based [14]. | Very Low – Typically requires a fixed laboratory setting; portable versions exist but are less common [15]. |
| Analysis Speed | Fast – Analysis times often between 3-5 minutes; rapid IMS spectrum acquisition in milliseconds enables real-time monitoring [14] [4]. | Moderate to Slow – Longer run times common; analysis can take weeks from sample collection to final report in forensic applications [15]. |
| Cost-Effectiveness | Higher – Lower initial investment and operational costs; uses air or nitrogen as drift gas, avoiding expensive helium [16] [17]. | Lower – High initial cost, substantial infrastructure needs, and ongoing consumables like scarce helium [16]. |
| Sensitivity | High – Detection limits in the mid parts-per-trillion (pptv) range without sample enrichment; IMS can be ~10x more sensitive than MS for some compounds [16] [18]. | High – Excellent sensitivity, but may require sample pre-concentration to achieve similar detection levels for trace analytes. |
| Operational Requirements | Simple – Operates at atmospheric pressure; no high vacuum required; minimal infrastructure [14]. | Complex – Requires high vacuum conditions; significant energy consumption; high-demand infrastructure [16]. |
| Linear Dynamic Range | Narrower – Typically linear over one order of magnitude before response becomes logarithmic, though linearization strategies can extend this [18]. | Broader – Maintains linearity over three orders of magnitude or more [18]. |
Performance Data and Experimental Evidence
Quantitative Performance and Stability
A comprehensive long-term study assessing a TD-GC-MS-IMS system provides robust data on the performance and stability of IMS detection [18].
Table 2: Long-Term Stability and Performance Data for GC-IMS
| Performance Metric | Result | Experimental Context |
|---|---|---|
| Long-Term Stability | Assessed over 16 months (156 measurement days) using ketones [18]. | Demonstrates instrument robustness for routine analysis. |
| Signal Intensity RSD | 3% to 13% (Relative Standard Deviation) [18]. | Indicates high measurement precision. |
| Retention Time Deviation | 0.10% to 0.22% [18]. | Shows excellent chromatographic stability. |
| Drift Time Deviation | 0.49% to 0.51% [18]. | Confirms high stability in the IMS dimension. |
| Sensitivity vs. MS | IMS was approximately ten times more sensitive than MS for the compounds studied [18]. | Limits of detection (LOD) in the picogram per tube range were achieved. |
Experimental Workflow: On-Site Analysis
The workflow for on-site analysis with portable GC-IMS differs significantly from traditional lab-based methods, contributing to its speed and efficiency. The following diagram illustrates a generalized protocol for rapid on-site screening using a portable GC-IMS system, incorporating elements from forensic and VOC analysis studies [18] [15].
Key Workflow Steps:
- Sample Collection: For volatile compounds, air or vapor is sampled directly, sometimes using concentrating devices like thermal desorption (TD) tubes [18] or Capillary Microextraction of Voliles (CMV) [15]. This step can be completed in minutes with minimal preparation.
- Sample Introduction: The sample is rapidly introduced into the system, often via thermal desorption, which releases analytes into the gas chromatograph [18].
- GC Separation: A fast GC method, potentially using low-thermal-mass (LTM) or multi-capillary columns (MCC), separates the complex mixture. Typical analysis times range from 3 to 5 minutes [14] [15].
- IMS Detection: Separated compounds enter the IMS drift tube, are ionized (e.g., by a low-dose radioactive source), and are separated based on their mobility in an electric field. This separation occurs on a millisecond timescale, generating a 2D spectrum (retention time vs. drift time) [4] [14].
- Data Analysis & Reporting: Data is processed using onboard software. Chemometric techniques like Principal Component Analysis (PCA) are often applied for complex sample discrimination, enabling immediate, on-site interpretation [16] [17].
Methodology and Reagent Solutions
Detailed Experimental Protocol
The following methodology is synthesized from studies that systematically evaluated GC-IMS performance, particularly for VOC analysis [18].
Aim: To quantify volatile organic compounds (VOCs) in a gas-phase sample using a GC-IMS system and compare its performance to GC-MS.
Materials and Equipment:
- GC-IMS instrument (portable or benchtop)
- Thermal Desorption (TD) unit with a controlled sampling system for TD tubes
- Standard compounds (e.g., ketones, aldehydes, alcohols) of high purity (≥95%)
- Methanol (GC-grade) as solvent for preparing calibration solutions
- Gas-tight syringes
- Data analysis software with chemometric capabilities
Procedure:
- Calibration Solution Preparation: Prepare separate stock solutions for different compound classes (e.g., alcohols, aldehydes, ketones) in methanol. Serially dilute to create a calibration series [18].
- Standard Loading: Load precise amounts of the calibration solutions onto the TD tubes using a controlled system that manages temperature and gas flow to ensure reproducible adsorption onto the sorbent material [18].
- Instrumental Analysis: Introduce the TD tube into the thermal desorber. The released analytes are carried to the GC column by a carrier gas (e.g., nitrogen or air). After chromatographic separation, analytes enter the IMS drift tube, which is maintained with a constant electric field and swept by a clean drift gas (e.g., nitrogen or purified air) [18] [4].
- Data Acquisition: For each analyte, the system records a 2D map of GC retention time and IMS drift time. Signal intensity, retention time, and drift time are tracked for quantitative and qualitative analysis [18] [14].
- Quantification and Linearization: Construct a calibration curve. Due to the narrow linear dynamic range of IMS (often ~1 order of magnitude), apply a linearization strategy to extend the usable calibration range to two orders of magnitude before the response becomes logarithmic [18].
Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for GC-IMS Analysis
| Item | Function/Description | Application Note |
|---|---|---|
| Thermal Desorption (TD) Tubes | Small tubes packed with adsorbent material to trap and pre-concentrate VOCs from air or gas samples [18]. | Enables standardized and reproducible sampling beyond the laboratory; critical for on-site analysis. |
| Drift Gases (N₂, Clean Air) | High-purity nitrogen or clean, dry air used as the buffer gas in the IMS drift tube [4]. | Operates at atmospheric pressure; avoids the use of scarce and expensive helium, reducing running costs [16]. |
| Chemical Standards | High-purity (>95%) reference substances for target analytes (e.g., ketones, aldehydes) used for calibration [18]. | Essential for compound identification and quantification, especially given the lack of universal IMS libraries. |
| Capillary Microextraction of Volatiles (CMV) | A dynamic headspace sampling device with a sol-gel adsorption phase for enhanced retention of volatiles like BTEX compounds [15]. | Used for rapid, on-site sampling and pre-concentration, coupling directly to portable GC-MS and GC-IMS systems. |
Critical Interpretation of Data
The data indicates that the choice between GC-IMS and traditional lab systems is application-dependent.
Strengths of GC-IMS: The technology is unequivocally superior for applications requiring rapid, on-site decisions. Its portability, speed (minutes versus hours or days), lower operational costs, and high sensitivity make it ideal for quality control in food processing, environmental monitoring, forensic triage at crime scenes, and clinical breath analysis [16] [17] [14]. Its untargeted analysis mode without sensitivity loss is a significant advantage for discovering unknown compounds [16].
Considerations and Limitations: GC-IMS has a narrower linear dynamic range compared to GC-MS, which can be a limitation for quantifying analytes across wide concentration ranges, though linearization strategies are being developed to mitigate this [18]. While excellent for targeted screening and pattern recognition, GC-IMS can face challenges with complex mixture ionization, where analytes may compete or suppress each other's signals [4]. Finally, the lack of a universal reference database for IMS spectra means identification often requires building in-house libraries or using parallel MS detection for confirmation [18] [14].
GC-IMS presents a compelling alternative to traditional lab-based systems like GC-MS, with distinct advantages in portability, speed, and cost-effectiveness. Experimental data confirms its high sensitivity, excellent long-term stability, and suitability for rapid on-site screening. While GC-MS remains the gold standard for definitive identification and applications requiring a wide linear dynamic range, GC-IMS has carved out a vital niche. It empowers researchers and professionals to perform high-quality, sensitive chemical analysis directly at the point of need, aligning with the growing demand for decentralized, green analytical technologies [16] [17].
Gas Chromatography-Ion Mobility Spectrometry (GC-IMS) represents a powerful two-dimensional analytical technique that combines the superior separation capabilities of gas chromatography with the rapid detection and characterization strengths of ion mobility spectrometry [4]. This hyphenated technique provides researchers with a robust tool for analyzing complex volatile organic compound (VOC) mixtures, delivering a two-dimensional spectrum where signal intensity is plotted as a function of GC retention time and IMS drift time [14]. The true analytical power of GC-IMS lies in the orthogonality of these two separation mechanisms—GC separates compounds based on their partitioning between stationary and mobile phases, while IMS separates ionized molecules based on their size, shape, and charge as they drift through a buffer gas under an electric field [4] [19].
The fundamental advantage of this orthogonal approach is the significantly enhanced peak capacity compared to either technique alone. While GC separation of complex mixtures might require analysis times of several minutes to hours, the IMS dimension operates on a millisecond timescale, allowing multiple mobility spectra to be captured across each chromatographic peak [4]. This combination has proven particularly valuable in applications requiring rapid analysis of complex samples, including food quality assessment [2] [20], biological sample analysis [18], and on-site environmental monitoring [19].
Fundamental Principles of 2D Data Generation
Gas Chromatography Separation Dimension
In the first separation dimension, gas chromatography operates on the principle of partitioning between a stationary phase (the coated inner wall of a capillary column) and a mobile phase (inert carrier gas). Compounds are separated based on their volatility and chemical affinity for the stationary phase, with different molecules eluting at characteristic retention times (RT) [4] [19]. For GC-IMS applications, both standard capillary columns and multi-capillary columns (MCC) are employed, with the latter being particularly advantageous for portable systems due to their higher sample capacity and faster analysis times [14]. The GC separation process typically occurs over a timeframe of several minutes, with modern systems achieving analysis times of 3-5 minutes for many applications [14].
Ion Mobility Spectrometry Separation Dimension
Following GC separation, eluting compounds enter the IMS ionization region, where they are typically ionized using a β-radiation source (such as ³Ni or ³H) [14] [21]. This ionization process occurs at atmospheric pressure through chemical ionization mechanisms, where reactant ions (often clustered water molecules) transfer charge to analyte molecules through proton or electron transfer reactions [4] [21]. The resulting ions are then pulsed into the drift tube through an ion shutter or gate [4].
Within the drift tube, ions migrate under the influence of a uniform electric field through a counter-flowing drift gas (typically nitrogen or purified air) [19]. Their velocity through this field depends on their collision cross-section—a function of the ion's size, shape, and mass—as well as the charge they carry [19]. This separation is characterized by the drift time (DT), which is related to the reduced ion mobility (K₀) through a standardized equation that normalizes for temperature and pressure conditions [19]. The IMS separation occurs remarkably quickly, with complete mobility spectra typically acquired in tens to hundreds of milliseconds [4].
Orthogonality in Separation Mechanisms
The power of GC-IMS stems from the fundamentally different physical principles governing each separation dimension. While GC separation depends primarily on thermodynamic properties (volatility and partitioning coefficients), IMS separation relies on electrophoretic mobility in the gas phase [4]. This orthogonality means that compounds with similar retention times may have distinctly different drift times, and vice versa, significantly enhancing the ability to resolve complex mixtures that would be challenging for either technique alone [4] [20].
Table 1: Comparison of Separation Mechanisms in GC-IMS
| Dimension | Separation Principle | Governing Factors | Timescale | Primary Outcome |
|---|---|---|---|---|
| GC Dimension | Partitioning between stationary and mobile phases | Volatility, polarity, molecular weight | Minutes | Retention time (RT) |
| IMS Dimension | Ion mobility in electric field | Collision cross-section, charge, mass | Milliseconds | Drift time (DT) |
Data Structure and Spectral Interpretation
The GC-IMS Spectrum Visualization
A GC-IMS spectrum is typically visualized as a two-dimensional contour plot or heat map, with the x-axis representing GC retention time, the y-axis representing IMS drift time, and signal intensity indicated by color intensity [4] [14]. Each detected compound appears as a spot or peak at specific RT/DT coordinates, creating a distinctive "fingerprint" for the sample being analyzed [22]. The appearance of these spectra has been likened to topographic maps, with mountains representing high signal intensity regions [22]. In the default visualization, the reactant ion peak (RIP)—resulting from the ionization of carrier gas molecules—typically appears as a prominent feature at a consistent location in the spectrum, serving as a valuable reference point [4].
Key Features and Patterns in 2D Spectra
When interpreting GC-IMS spectra, several characteristic patterns provide important chemical information:
Monomer and Dimer Formation: Many compounds appear as multiple spots vertically aligned at the same retention time but different drift times, representing monomer and dimer ions [14]. The relative abundance of these forms is concentration-dependent, with dimers typically becoming more prominent at higher concentrations [14].
Isomer Separation: Isomeric compounds with identical mass but different structures often have similar retention times but different drift times due to variations in their collision cross-sections, allowing for their differentiation [14] [20].
Matrix Effects: Changes in background composition (humidity, competing analytes) can cause signal suppression or mobility shifts, highlighting the importance of consistent sample preparation and analysis conditions [4].
The following diagram illustrates the typical workflow for generating and interpreting a GC-IMS spectrum:
Experimental Protocols for GC-IMS Analysis
Proper sample preparation is critical for reproducible GC-IMS results. For solid or liquid samples, headspace sampling is the most common approach, where volatile compounds are allowed to equilibrate in the gas phase above the sample in a sealed vial [2] [20]. The headspace volume is then injected into the GC-IMS system using a gas-tight syringe or automated sampling system. For trace analysis, pre-concentration techniques such as thermal desorption tubes or solid-phase microextraction (SPME) may be employed to enhance sensitivity [18] [21]. When analyzing aqueous samples, humidity control is essential, as water vapor can significantly impact IMS ionization efficiency and mobility measurements [4] [18].
Instrument Configuration and Method Parameters
Standard GC-IMS methodology involves careful optimization of both GC and IMS parameters:
GC Conditions: Column selection (typically weakly polar stationary phases for volatile compounds), column temperature (isothermal or programmed), and carrier gas flow rate (balanced to minimize peak broadening while maintaining reasonable analysis times) [4] [19].
IMS Conditions: Drift tube temperature (typically 45-60°C), drift gas flow rate (usually 150-300 mL/min), and electric field strength (typically 200-400 V/cm) [19] [20].
Data Acquisition: Rapid spectral acquisition rate (typically 10-100 spectra/second) to ensure sufficient data points across both GC and IMS peaks [4].
The following table outlines typical experimental parameters for food flavor analysis using headspace GC-IMS:
Table 2: Standard GC-IMS Parameters for Food Flavor Analysis [2] [20]
| Parameter | GC Conditions | IMS Conditions | Sample Preparation |
|---|---|---|---|
| Column Type | FS-SE-54-CB-1 (15 m × 0.53 mm) | Drift tube length: 5-10 cm | Headspace incubation: 15 min |
| Column Temperature | 60°C (isothermal) | Drift tube temperature: 45°C | Incubation temperature: 60°C |
| Carrier Gas | Nitrogen | Drift gas: Nitrogen | Sample amount: 2 g |
| Flow Rate | 2 mL/min initial, ramping to 100 mL/min | 150 mL/min | Injection volume: 500 μL |
| Analysis Time | 20 minutes | Spectrum acquisition rate: 10-50 Hz | - |
Data Processing and Peak Detection Algorithms
Raw GC-IMS data requires specialized processing to extract meaningful information. Recent advances include the application of topological data analysis (TDA) and persistent homology for automated 2D peak detection [22]. These algorithms naturally identify significant features (peaks) in the 2D data and measure their persistence (significance), enabling reliable chemical identification and quantification [22]. Standard processing workflows typically include:
- Baseline correction to remove background effects
- Noise reduction algorithms to improve signal-to-noise ratio
- Peak alignment to correct for minor retention time and drift time shifts between runs
- Peak picking and integration for quantification [22]
Comparative Performance Analysis
GC-IMS vs. GC-MS: Analytical Capabilities
When compared to the established gold standard of GC-MS, GC-IMS demonstrates distinct advantages and limitations:
Table 3: Performance Comparison: GC-IMS vs. GC-MS [18] [20]
| Parameter | GC-IMS | GC-MS | Practical Implications |
|---|---|---|---|
| Sensitivity | ~10x higher for certain compounds [18] | Excellent | IMS better for trace volatile analysis |
| Linear Range | 1-2 orders of magnitude [18] | 3+ orders of magnitude [18] | MS superior for quantification across wide concentration range |
| Isomer Separation | Excellent (based on structural differences) | Limited (similar fragmentation patterns) | IMS advantageous for isomeric compounds |
| Identification Capability | Limited databases, requires standards | Extensive spectral libraries | MS better for unknown identification |
| Analysis Environment | Ambient pressure operation | High vacuum required | IMS more suitable for portable, field-deployable instruments |
| Speed of Analysis | Very fast (seconds to minutes) | Moderate to fast | IMS better for high-throughput screening |
| Instrument Cost | Lower | Higher | IMS more accessible for routine analysis |
Quantitative Performance and Long-Term Stability
Recent systematic evaluation of GC-IMS quantification performance demonstrates excellent long-term stability, with relative standard deviations for signal intensities ranging from 3% to 13% over 16 months and 156 measurement days [18]. Retention time deviations remained minimal (0.10-0.22%), while drift time variations were slightly higher but still acceptable (0.49-0.51%) [18]. This remarkable stability makes GC-IMS suitable for long-term monitoring applications and quality control processes where consistent performance is essential.
For quantitative applications, IMS response typically shows linear behavior over approximately one order of magnitude (e.g., 0.1 to 1 ng/tube for pentanal) before transitioning to a logarithmic response at higher concentrations [18]. Through linearization strategies, this usable calibration range can be extended to approximately two orders of magnitude [18]. This compares to GC-MS, which typically maintains linearity over three orders of magnitude (up to 1000 ng/tube) [18].
Essential Research Tools and Reagents
Successful GC-IMS analysis requires specific reagents and consumables optimized for the technique:
Table 4: Essential Research Reagent Solutions for GC-IMS
| Reagent/Consumable | Function/Purpose | Technical Specifications | Application Notes |
|---|---|---|---|
| Drift Gases | Inert buffer gas for IMS separation | High-purity nitrogen or dried air (dew point < -90°C) [19] | Requires moisture trap to prevent humidity effects [4] |
| Calibration Standards | System calibration and compound identification | Ketones (e.g., acetone, 2-butanone) or external calibration mixtures [18] | Regular calibration essential for reproducible drift times [18] |
| GC Columns | Primary separation dimension | Multi-capillary columns (MCC) or wide-bore capillary columns (0.53 mm ID) [4] [14] | MCC provides higher sample capacity for complex mixtures [4] |
| Thermal Desorption Tubes | Sample pre-concentration for trace analysis | Tubes packed with Tenax TA, Carbograph, or other sorbents [18] | Enables detection of pptv-level compounds [18] |
| Internal Standards | Signal normalization and quantification | Deuterated compounds or stable isotope-labeled analogs | Corrects for injection volume variations and signal drift |
Applications in On-Site Screening vs. Laboratory Analysis
The unique capabilities of GC-IMS make it particularly valuable for specific application scenarios:
Field-Deployable GC-IMS Systems
Miniaturized GC-IMS instruments have been developed for hand-held operation, featuring dramatically reduced dimensions (e.g., 170 mm × 110 mm × 55 mm) while maintaining impressive performance characteristics [19]. These portable systems achieve detection limits in the parts-per-trillion (pptv) range with analysis times of just 125 milliseconds for IMS averaging [19]. Such capabilities enable real-time monitoring applications in security (explosives and chemical warfare agent detection), environmental monitoring (air quality assessment), and industrial process control [19] [21].
Laboratory-Based Applications
In laboratory settings, GC-IMS has found particular utility in food quality assessment [2] [20], breath analysis for medical diagnostics [18], and metabolomics studies [20]. The technique's high sensitivity to volatile flavor compounds has made it invaluable for discrimination of food authenticity, monitoring flavor changes during storage and processing, and detection of microbial contamination [2]. In combined GC-IMS and GC-MS approaches, researchers can leverage the complementary strengths of both techniques for comprehensive volatile compound profiling [20].
GC-IMS represents a powerful analytical technique that leverages orthogonal separation mechanisms to provide enhanced resolution of complex volatile mixtures. Its strengths in sensitivity, speed of analysis, and portability make it particularly valuable for applications requiring rapid screening and field analysis. While limitations in compound identification and quantitative linear range remain compared to GC-MS, ongoing advances in instrumentation, data processing algorithms, and standardized methodologies continue to expand its applications across diverse fields. As database libraries grow and hyphenated systems become more sophisticated, GC-IMS is poised to become an increasingly indispensable tool in the analytical scientist's arsenal, particularly for scenarios where speed, sensitivity, and portability are paramount.
GC-IMS in Action: Protocols and Real-World Screening Applications
The field of analytical chemistry is witnessing a paradigm shift from centralized laboratory analysis to rapid, on-site screening techniques. This transition is particularly evident in applications requiring immediate results, such as environmental monitoring, food safety controls, and clinical diagnostics. Gas Chromatography coupled with Ion Mobility Spectrometry (GC-IMS) has emerged as a powerful technology bridging this gap, offering laboratory-grade separations with the portability and speed necessary for field deployment [16]. The analytical performance of any GC-IMS system, however, is fundamentally dependent on the initial sampling technique employed to introduce analytes into the system. This guide provides a comprehensive comparison of three fundamental on-site sampling approaches—headspace, thermal desorption, and swab analysis—framed within the context of optimized workflows for GC-IMS. We objectively evaluate their performance characteristics, supported by experimental data, to inform researchers and development professionals in selecting the most appropriate methodology for their specific application requirements.
Core Sampling Techniques: Principles and Applications
Headspace Sampling
Principle: Headspace sampling involves heating a sample in a sealed vial to equilibrium, allowing volatile organic compounds (VOCs) to partition into the gas phase above the sample (the headspace). A portion of this headspace gas is then injected into the GC-IMS for analysis [23].
Advantages and Limitations: This technique is most suited for the analysis of very light volatiles and is ideal for automated, high-throughput screening applications [23]. Its primary limitation is sensitivity, typically confined to the ppm to ppt range, and it is less effective for higher-boiling-point semi-volatiles that do not efficiently partition into the gas phase [23]. A notable advancement is Dynamic Headspace (DHS), which continuously sweeps the headspace with an inert gas through an adsorbent trap, concentrating the analytes and providing lower detection limits and reduced water interference compared to traditional static headspace or Purge and Trap methods [24].
Thermal Desorption-Based Techniques
Principle: This category encompasses techniques that release and transfer analytes from a sample through the application of heat. Two prominent methods are:
- Purge and Trap Thermal Desorption (P&T): An inert gas is bubbled through a liquid sample (or purges a solid), "stripping" volatiles which are then concentrated on an adsorbent trap. The trap is subsequently heated to thermally desorb the analytes into the GC-IMS [23] [24]. While sensitive (ppb range), it can be time-consuming and may miss very light volatiles that break through the trap [23].
- Direct Thermal Extraction (DTE): Solid or liquid samples are placed directly into a desorption tube and rapidly heated, volatilizing a wide range of compounds for immediate transfer to the GC-IMS. This technique requires virtually no sample preparation and offers high sensitivity for a broad boiling-point range of organics [23] [25].
Swab Sampling
Principle: Swab analysis is a surface sampling technique where a swab tip, often moistened with a solvent, is wiped across a surface to collect analytes. The swab is then typically placed into a thermal desorption unit or extracted with a solvent to introduce the collected compounds into the analytical instrument [26].
Advantages and Limitations: This method is exceptionally simple, non-destructive, and ideal for on-site, targeted analysis of specific surfaces, such as pesticide residues on fruit or contaminants on equipment [26]. Its primary strength lies in its ability to perform rapid, in-situ sampling with minimal equipment, making it a perfect companion for portable GC-IMS systems.
Comparative Performance Evaluation
The selection of a sampling technique is a critical determinant of analytical success. The table below summarizes the key performance characteristics of the discussed methods, providing a basis for objective comparison.
Table 1: Comparative Performance of On-Site Sampling Techniques for VOC Analysis
| Parameter | Headspace GC | Purge & Trap TD | Direct Thermal Extraction (DTE) | Swab Analysis |
|---|---|---|---|---|
| Typical Sensitivity | ppm to ppt range [23] | ppb range [23] | ppb range; 10-1000x more sensitive than P&T/HS [23] [25] | High (varies with analyte/desorption) [26] |
| Optimal Analyte Range | Very light volatiles [23] | Volatiles, some semi-volatiles [23] | Wide range of volatiles and semi-volatiles [23] | Surface residues, non-volatiles (with ESI) [26] |
| Sample Preparation | Minimal | Moderate | Minimal to none [23] | Minimal |
| Water Interference | Low | High (requires water management) [23] | High (problematic for moist samples) [23] | Low (method-dependent) |
| Throughput/Automation | Excellent [23] | Moderate | Good | Good |
| Best For | Screening, gases, automated QC | Environmental waters, regulated methods | Solids, viscous liquids, broad profiling | Surface analysis, forensic, food safety [26] |
Table 2: Experimental Data from Comparative Studies [23] [25]
| Sample Matrix | Headspace GC Results | Purge & Trap TD Results | Direct Thermal Extraction (DTE) Results |
|---|---|---|---|
| Olive Oil | Only two minor peaks detected from 5.0 mL [23]. | Sensitive analysis from 1.0 mL [23]. | 100x more sensitive than P&T; 1000x more sensitive than HS; detected wider boiling point range from only 10.0 µL [23] [25]. |
| Gasoline in Water | Useful for very volatile analytes and aromatics; less sensitive for heavier compounds [23]. | ~1000x more sensitive than HS for less volatile aromatics and semi-volatiles [23]. | Not tested in this specific experiment. |
| Black Tea in Water | No significant peaks detected [23]. | Effective analysis [23]. | More sensitive than P&T; detected a wider range of compounds [23]. |
Workflow Integration with GC-IMS
GC-IMS serves as an ideal detection platform for on-site analysis due to its high sensitivity, portability, and operation at atmospheric pressure [16] [4]. The coupling of sampling techniques with GC-IMS creates a powerful, two-dimensional separation (GC retention time and IMS drift time) that effectively deconvolutes complex mixtures [4]. The following diagram illustrates the logical workflow for method selection and analysis.
Detailed Experimental Protocols
Protocol: Headspace GC-IMS for Microbial VOC Profiling
This protocol, adapted from research on identifying wound infection bacteria, details the detection of microbial volatile organic compounds (mVOCs) [27].
- Sample Preparation: Inoculate 1.5 mL of thioglycolate medium in a 20 mL sampling bottle with the target microorganism (e.g., Escherichia coli, Staphylococcus aureus). Incubate at 37°C for 12-15 hours without agitation.
- Instrument Parameters:
- GC-IMS System: Configured with a wide-bore GC column (e.g., mxt-5, 15 m × 0.53 mm).
- Incubation: 60.0 °C for 10.0 min with shaking.
- Injection: 1 mL of headspace gas.
- GC Temperature: 40 °C.
- Drift Tube Temperature: 45 °C.
- Carrier/Drift Gas: Nitrogen.
- Analysis Time: 25 min [27].
- Key Findings: The study successfully differentiated between single and mixed cultures of bacteria based on their unique mVOC fingerprints, demonstrating the utility of headspace GC-IMS for rapid microbiological identification [27].
Protocol: Swab-ESI-IMS for Pesticide Residue Detection
This protocol describes a non-destructive method for detecting pesticide residues on fruit surfaces using swab sampling coupled with Electrospray Ionization-Ion Mobility Spectrometry (ESI-IMS) [26].
- Swab Collection: Manually swab a defined area (e.g., 5x5 cm) on the fruit's surface (e.g., apple, pear). A pre-moistened (with methanol) swab can be used to improve recovery.
- Sample Extraction: Place the swab tip into a vial containing 1 mL of elution solvent (e.g., methanol). Vortex for a set time (e.g., 1-2 minutes) to release the pesticides into the solution.
- ESI-IMS Analysis:
- Ionization: Electrospray Ionization (ESI) source, suitable for non-volatile pesticides.
- Optimized Parameters: ESI bias voltage: ~3.5 kV; Drift tube temperature: ~120 °C; Solution flow rate: ~20 μL/min [26].
- Direct Injection: The extracted solution is directly injected into the ESI-IMS, bypassing the need for chromatographic separation for rapid screening.
- Key Findings: The method showed wide linear range, high sensitivity, and acceptable repeatability for pesticides like carbendazim and thiabendazole. Recovery rates in spiked fruit samples ranged from 71.4 to 121.2% [26].
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Materials and Reagents for On-Site Sampling Workflows
| Item | Function/Application | Notes |
|---|---|---|
| Tenax TA Adsorbent | Trapping material for volatile organics in DTE, P&T, and DHS workflows. | High thermal stability; hydrophobic, minimizes water retention [23]. |
| Tritium (³H) Ionization Source | Standard ionization source inside the IMS drift tube. | Ionizes gas molecules for detection; low-dose sealed sources are common in modern GC-IMS [16]. |
| GC Cryo-Trap | Focuses volatiles at the GC column head after thermal desorption. | Prevents band broadening by cryo-focusing analytes at low temps (e.g., -100 °C) before analysis [23]. |
| Sampling Vials/Bottles | Contain liquid or solid samples for headspace or purge and trap analysis. | Must be sealable (with crimp or screw caps) and withstand heating (e.g., 20 mL vials) [27]. |
| Surface Swabs | Collection of analytes from solid surfaces for subsequent analysis. | Typically cotton or synthetic fiber; can be pre-moistened for improved recovery of residues [26]. |
| Nitrogen Generator | Provides high-purity carrier and drift gas for GC-IMS operation. | Enables instrument operation without gas cylinders, enhancing portability for on-site use [27]. |
The optimization of on-site sampling workflows is paramount for unlocking the full potential of GC-IMS technology. As demonstrated, the choice of sampling technique—headspace, thermal desorption, or swab analysis—profoundly impacts sensitivity, analyte range, and applicability. No single method is universally optimal; the selection must be driven by the sample matrix, the physicochemical properties of the target analytes, and the required speed of analysis. Direct Thermal Extraction offers superior sensitivity for solid samples, while Dynamic Headspace provides robust, automated analysis of liquids with minimal water interference. Swab analysis remains the unequivocal choice for surface monitoring. By integrating these optimized sampling protocols with the portability and analytical power of GC-IMS, researchers and professionals can perform sophisticated, laboratory-grade analysis directly in the field, dramatically accelerating decision-making across drug development, food safety, and environmental monitoring.
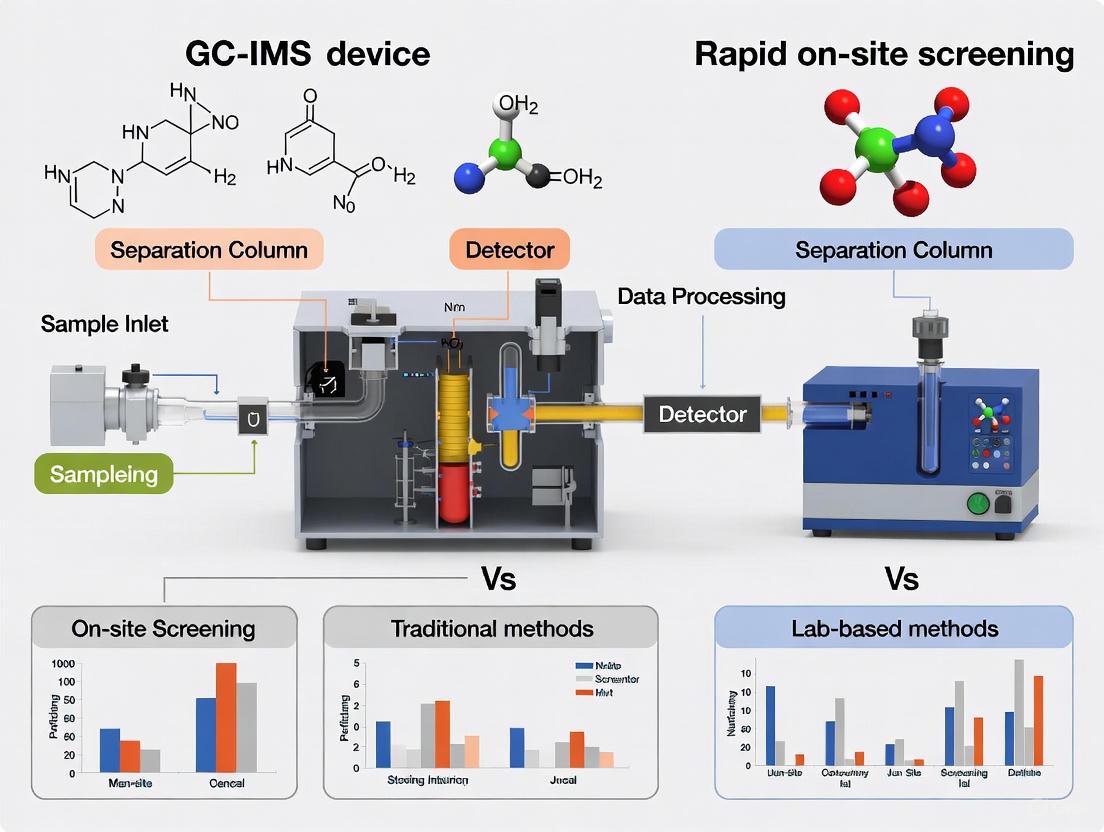
The illegal production of synthetic drugs, particularly methamphetamine, in clandestine laboratories presents an emerging global threat. Locating these hidden facilities is a critical challenge for law enforcement agencies, necessitating analytical methods that can reliably detect trace amounts of precursor chemicals in the gas phase at these sites. While traditional laboratory techniques like gas chromatography-mass spectrometry (GC-MS) offer high sensitivity, they lack the portability and speed required for on-site screening. In this context, Gas Chromatography-Ion Mobility Spectrometry (GC-IMS) has emerged as a powerful technology that balances high sensitivity with field-deployable capabilities. This technology enables the detection of relevant methamphetamine synthesis chemicals at concentrations as low as the single-digit parts per billion by volume (ppbv) range, providing law enforcement with a viable tool for locating illicit drug production facilities [12].
The fundamental advantage of GC-IMS lies in its dual-separation approach, which combines the separation power of gas chromatography with the rapid detection capabilities of ion mobility spectrometry. This combination is particularly suited for targeted analysis of predefined compounds in complex samples, enhancing quantitative performance by reducing chemical noise and enriching analyte signals [28]. For methamphetamine precursor detection, this means reliable identification of target compounds even in the presence of complex background interferences commonly found in environments where clandestine labs operate.
Fundamental Principles
GC-IMS operates through a two-stage separation process that provides complementary information for compound identification. In the first stage, the gas chromatography (GC) component separates volatile organic compounds (VOCs) based on their partitioning between a mobile gas phase and a stationary liquid phase coated on the chromatographic column wall. This separation depends on the compounds' vapor pressures and interactions with the stationary phase, with different chemicals eluting at characteristic retention times [29].
Following GC separation, the ion mobility spectrometry (IMS) stage further separates ions based on their size, charge, and shape as they drift through a buffer gas under the influence of an electric field. The drift time is influenced by the ion's collision cross section (CCS) - a measurable physical property that adds a complementary identification parameter alongside GC retention time. The final detection occurs when these separated ions strike a Faraday plate, generating a signal [29]. The combination of retention time from GC and drift time from IMS creates a two-dimensional dataset that enables highly specific compound identification while minimizing false positives from cross-sensitivities [12].
Key Technological Advantages
- High Sensitivity: Capable of detecting target analytes at single-digit ppbv levels [12]
- Rapid Analysis: Provides results within seconds to minutes, enabling real-time decision making
- Portability: Compact designs allow for field deployment in various operational scenarios
- Operational Simplicity: Requires minimal sample preparation compared to laboratory-based techniques
- Robustness: Functions reliably in various environmental conditions without complex maintenance
Experimental Performance Data: Detection of Methamphetamine Precursors
Recent research has demonstrated the practical capabilities of GC-IMS for detecting methamphetamine synthesis precursors. A comprehensive study investigated the feasibility of detecting precursors involved in the three main synthesis pathways of benzyl methyl ketone (BMK) - the Dakin-West method, the nitrostyrene method, and the Baeyer-Villinger pathway. The results confirmed that GC-IMS can reliably detect these target compounds at single-digit ppbv concentrations based on their specific retention times and reduced ion mobility values [12].
Table 1: Detection Capabilities of GC-IMS for Methamphetamine Synthesis Pathway Chemicals
| Synthesis Pathway | Target Precursors | Detection Limit (ppbv) | Identification Parameters |
|---|---|---|---|
| Dakin-West Method | Specific precursor compounds | Single-digit range | Retention time + Reduced ion mobility |
| Nitrostyrene Method | Specific precursor compounds | Single-digit range | Retention time + Reduced ion mobility |
| Baeyer-Villinger Pathway | Specific precursor compounds | Single-digit range | Retention time + Reduced ion mobility |
| Real Seized Samples | BMK and synthesis by-products | Demonstrated capability | Retention time + Reduced ion mobility |
The practical utility of this approach was further validated using a real seized sample of BMK, where GC-IMS successfully detected characteristic by-products in the headspace, enabling potential drug profiling through gas phase sampling near suspicious premises [12]. This capability is particularly valuable for law enforcement operations where non-invasive screening of facilities is required before pursuing search warrants.
Comparison with Alternative Analytical Techniques
Laboratory-Based Methods
Traditional laboratory methods for drug precursor analysis include Gas Chromatography-Mass Spectrometry (GC-MS) and High-Performance Liquid Chromatography (HPLC). While these techniques offer excellent sensitivity and identification capabilities, they present significant limitations for field deployment.
Table 2: Comparison of GC-IMS with Laboratory-Based Analytical Techniques
| Parameter | GC-IMS | GC-MS | HPLC |
|---|---|---|---|
| Detection Sensitivity | Single-digit ppbv range [12] | Similar or slightly better | ~mg/mL range [30] |
| Analysis Time | Seconds to minutes | Minutes to hours | 10-30 minutes [30] |
| Portability | High - field deployable | Low - laboratory bound | Low - laboratory bound |
| Sample Preparation | Minimal - often direct gas sampling | Extensive - may require derivatization | Moderate - extraction needed [30] |
| Operational Complexity | Low - minimal training required | High - requires expert operators | Moderate - requires trained technicians |
| Identification Power | Two-dimensional (retention + mobility) | Two-dimensional (retention + mass spectrum) | Primarily retention time based |
| Cost of Ownership | Moderate | High | Moderate to High |
GC-MS traditionally provides superior identification power through mass spectral matching but requires complex maintenance and highly trained personnel [31]. HPLC methods, while effective for quantifying methamphetamine in solid samples with good linearity (R² = 0.9999) and precision (RSD = 2.9%), lack the volatility-based separation needed for direct gas phase analysis of precursors [30].
Other Field-Deployable Techniques
Other portable detection technologies include standalone IMS devices and optical spectroscopy methods. While standalone IMS offers extreme portability and rapid response, it suffers from lower resolving power (often less than 100) and significant susceptibility to ion suppression effects in complex sample matrices [32]. These limitations can lead to both false-positive and false-negative results in field applications.
Recent advancements in hyphenated systems show promise for bridging this performance gap. The coupling of ultrafast gas chromatographs with tandem differential mobility spectrometers (DMS-DMS) has demonstrated the ability to separate complex mixtures of nitroaromatic explosives in under 20 seconds while effectively mitigating in-source ion suppression effects through chromatographic separation prior to ionization [32]. This approach illustrates the growing trend toward hybrid instruments that balance analytical performance with field practicality.
Experimental Protocols for GC-IMS Analysis
Standard Methodology for Precursor Detection
Based on published research, the following protocol outlines a standardized approach for detecting methamphetamine precursors using GC-IMS:
Sample Collection and Introduction:
- Utilize passive headspace sampling or active air sampling techniques
- Collect gas phase samples in proximity to suspected synthesis areas
- Introduce samples via split/splitless inlet system with controlled split ratios (e.g., 20:1)
- Maintain inlet temperatures between 180-205°C depending on target compound volatility
Chromatographic Separation:
- Employ mid-polarity capillary columns (e.g., Rxi-5Sil MS, 2-2.2 m length)
- Utilize optimized temperature programs with rapid ramp rates (5-25°C/s)
- Implement carrier gas flows of 0.25-0.5 mL/min (nitrogen or purified air)
- Achieve separation of complex mixtures within 20-130 second timeframes
Ionization and Mobility Separation:
- Employ tritium or corona discharge ionization sources
- Utilize drift tubes with electric fields of 200-500 V/cm
- Implement drift gas flows (nitrogen) counter-current to ion motion
- Maintain drift tube temperatures between 40-100°C depending on application
Detection and Data Analysis:
- Monitor ion currents at Faraday plate detectors
- Process two-dimensional data (retention time vs. drift time)
- Identify compounds based on calibrated retention indices and reduced mobility values
- Quantify using peak volume or height comparisons with reference standards
Validation Procedures
Method validation should include:
- Determination of detection limits for target precursors using serial dilution
- Assessment of reproducibility through repeated measurements
- Evaluation of matrix effects by analyzing samples with potential interferents
- Verification of identification confidence through confirmation with certified reference materials
GC-IMS Experimental Workflow
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for GC-IMS Methamphetamine Precursor Analysis
| Item Category | Specific Examples | Function/Purpose |
|---|---|---|
| Reference Standards | BMK, P2P, phenylacetone, synthesis precursors | Method calibration and compound identification |
| Chromatographic Consumables | Rxi-5Sil MS columns (2-2.2 m × 0.1 mm × 0.1 μm), methyl deactivated transfer lines, SilTite Mini Union connectors | Compound separation and system connectivity |
| Gases and Carriers | High-purity nitrogen (carrier and drift gas), hydrogen (for FID detection when used), zero air | Mobile phase and detector operation |
| Sample Collection Materials | Sorbent tubes, gas sampling bags, passive diffusion samplers | Field sample acquisition and preservation |
| Quality Control Materials | Certified reference materials, internal standards, proficiency test samples | Method validation and quality assurance |
| Instrument Calibration | Reactive compounds (ketones, alcohols), n-ketone series for IMS calibration | System performance verification and mobility scale calibration |
Signaling Pathways and Detection Logic
The identification of methamphetamine synthesis pathways through VOC analysis relies on detecting characteristic chemical signatures associated with specific synthetic routes. The three primary pathways for BMK synthesis each generate distinct by-products and intermediate compounds that can be detected in the gas phase.
Pathway Identification Logic
GC-IMS technology represents a significant advancement in the rapid detection of methamphetamine synthesis precursors at clinically relevant single-digit ppbv concentrations. The technique successfully balances the competing demands of analytical sensitivity, identification certainty, and operational practicality that are essential for effective clandestine laboratory detection. While laboratory-based methods like GC-MS and HPLC provide superior identification power and sensitivity for confirmatory analysis, GC-IMS offers unmatched capabilities for rapid on-site screening that enables law enforcement to identify potential synthesis locations through non-invasive atmospheric sampling.
The dual-separation principle of GC-IMS, combining chromatographic retention with ion mobility, provides a robust framework for minimizing false positives while maintaining the portability and ease of use required for field deployment. As advances in miniaturization and data processing continue, GC-IMS platforms are poised to become increasingly valuable tools in the ongoing effort to combat illicit drug manufacturing, potentially integrating with wider chemical surveillance networks to provide comprehensive monitoring of precursor chemical diversion.
In the evolving landscape of analytical chemistry, Gas Chromatography-Ion Mobility Spectrometry (GC-IMS) is emerging as a powerful technology that bridges the critical gap between laboratory-based gold-standard methods and the pressing need for rapid, on-site screening. This technique combines the high separation capability of gas chromatography (GC) with the high sensitivity and portability of ion mobility spectrometry (IMS), enabling the accurate characterization of volatile organic compounds (VOCs) with minimal sample preparation [2]. While traditional methods like GC-Mass Spectrometry (GC-MS) remain the reference standard for definitive compound identification, they are typically time-consuming, require complex sample pretreatment, and are not suitable for field deployment [2]. In contrast, GC-IMS provides a viable alternative for non-destructive, rapid analysis, achieving detection at the parts per billion (ppb) level within minutes, making it particularly valuable for applications requiring immediate results [33] [2]. This guide objectively compares the performance of GC-IMS with other analytical techniques, focusing on its growing applications in food safety, specifically mold detection in maize, and clinical VOC profiling, framing its use within the broader thesis of rapid on-site screening versus conventional lab-based analysis.
Performance Comparison: GC-IMS vs. Alternative Analytical Techniques
The selection of an analytical technique involves balancing factors such as speed, sensitivity, portability, and analytical depth. The table below provides a structured comparison of GC-IMS against other common technologies used for VOC analysis.
Table 1: Performance Comparison of VOC Analysis Techniques
| Feature | GC-IMS | GC-MS | Electronic Nose (E-nose) | MALDI-TOF MS |
|---|---|---|---|---|
| Analysis Speed | Rapid (10-30 min) [33] [34] | Slow (can be hours) [2] | Very Rapid (minutes) [2] | Rapid (minutes for analysis) [35] |
| Sensitivity | High (ppb level) [33] | High (ppb/ppt level) | Variable, can be high [2] | High (for proteins) [35] |
| On-Site/Portable Use | Yes, portable systems available [2] | Limited; benchtop or large portable units [36] | Yes [2] | No, typically a large benchtop system [35] |
| Sample Preparation | Minimal (often none) [33] [34] | Complex (often requires extraction/pre-concentration) [37] [36] | Minimal | Extensive (mixing with matrix, partial cell lysis) [35] |
| Compound Identification | Library-based (NIST/IMS); can struggle with unknowns [34] | Definitive (mass spectrum library matching) | Pattern recognition only; no compound ID [2] | Library-based (spectral fingerprint matching) [35] |
| Key Advantage | Good separation & sensitivity with portability | Gold standard for definitive identification | Very fast and cheap for pattern recognition | Excellent for microorganism identification |
| Typical Application | Food spoilage, clinical VOC screening [2] [34] | Regulatory analysis, metabolite identification [2] | Quality control, spoilage screening [2] | Microbial identification in clinical labs [35] |
Application in Food Safety: Early Mold Detection in Maize
Experimental Protocol for Maize Mold Analysis
The application of GC-IMS for the early detection of mold in stored maize demonstrates its practical utility in food safety. A typical protocol, as outlined in recent research, involves the following steps [37]:
- Sample Preparation: Whole maize kernels (e.g., 1.0 g) are placed directly into a headspace vial and sealed. This non-destructive preparation is a key advantage.
- Incubation: The sealed vial is incubated at an elevated temperature (e.g., 80°C) for a short period (e.g., 15 minutes) with agitation to accelerate the release of VOCs into the headspace.
- GC-IMS Analysis:
- Injection: A specific volume (e.g., 500-1000 µL) of the headspace gas is automatically injected into the GC-IMS system.
- GC Separation: The VOCs are pre-separated using a weakly polar or polar capillary column (e.g., MXT-5 or MXT-WAX) with a temperature of 60-80°C. The carrier gas (N₂) flow is often programmed to ramp from low (e.g., 2 mL/min) to high (e.g., 100 mL/min) to separate compounds of varying volatilities.
- IMS Detection: The separated compounds enter the drift tube of the IMS, are ionized by a tritium source, and are separated based on their size, shape, and charge as they drift against a counter-flow of drift gas (N₂).
- Data Processing: The resulting 3D data (retention time, drift time, intensity) is processed using specialized software (e.g., VOCal, G.A.S.). Fingerprints are visualized in 2D topographic plots, and multivariate statistics like Principal Component Analysis (PCA) are used to classify samples.
Key Findings and Supporting Data
Research has successfully identified several volatile compounds as reliable early-warning biomarkers for initial mold development in maize, often before visible growth occurs [37]. The following table summarizes key biomarkers and the performance of the GC-IMS method.
Table 2: GC-IMS Performance in Maize Mold Detection
| Parameter | Findings |
|---|---|
| Key Biomarkers Identified | Propan-1-ol, Hexanal, Propanal, Nonanal, Butan-2-one, Isobutanol [37] |
| Classification Capability | Maize samples were categorized into four distinct stages of mold severity [37] |
| Validation Method | GC-IMS classification showed high consistency with traditional quality parameters (fatty acid value, total plate count) [37] |
| Early Warning | Four compounds (2-methyl-1-butanol, 2-propanone, 1-penten-3-ol) showed 1.3-1.5-fold increases in early mold stages in a similar study on tobacco [38] |
The workflow below summarizes the end-to-end process for GC-IMS analysis of mold in agricultural products.
Application in Clinical Diagnostics: VOC Profiling for Disease Detection
Experimental Protocol for Clinical VOC Analysis
In clinical settings, GC-IMS is being explored for non-invasive disease diagnosis through the analysis of VOCs in various bio-specimens. A study on oral cancer (OC) detection provides a robust protocol for collecting and analyzing different sample types [39]:
- Patient Preparation: Participants fast and abstain from smoking, alcohol, and personal care products for a specified period to minimize confounding VOCs.
- Sample Collection (Multiple Matrices):
- Exhaled Breath: Collected using a Haldane tube or device like the BioVOC-2 after a period of nasal breathing and a breath hold.
- Lesional Air: Air from directly next to a lesion in the mouth is drawn using a syringe or specialized device.
- Lesional Brushings: A soft cytology brush is used to collect cells from the lesion site, which is then placed in a headspace vial.
- Sample Introduction:
- Gaseous Samples (Breath/Lesional Air): Often introduced directly into the GC-IMS via a syringe or transferred to and expelled through a thermal desorption tube for later analysis.
- Solid/Liquid Samples (Brushings/Tissue): The headspace vial is incubated, and the VOCs are transferred to a thermal desorption tube using a device like a micro-chamber/thermal extractor.
- GC-IMS Analysis: The general analytical principles remain the same as for food analysis, focusing on detecting the subtle differences in VOC profiles between diseased and healthy groups.
Key Findings and Supporting Data
Clinical studies have demonstrated the strong potential of GC-IMS. In the oral cancer study, lesional brushings provided the best separation between OC patients and controls, followed by lesional air and exhaled breath when analyzed by TD-GC-MS, with GC-IMS also showing diagnostic value [39]. Key discriminatory compounds included various alkanes, alkenes, aromatic hydrocarbons, and ketones [39].
Furthermore, GC-IMS has been applied to identify bacteria and even their antibiotic resistance. A study on Escherichia coli successfully differentiated between carbapenem-sensitive (CSEC) and carbapenem-resistant (CREC) strains based on their volatile metabolic profiles, detecting 36 VOCs including alcohols, aldehydes, and ketones [34]. This highlights its potential for rapid antibiotic susceptibility testing, which could significantly improve patient prognosis.
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of GC-IMS experiments requires specific reagents and materials. The following table details key items and their functions based on the protocols from the search results.
Table 3: Essential Reagents and Materials for GC-IMS Experiments
| Reagent/Material | Function / Application | Example from Literature |
|---|---|---|
| Chemical Standards | Used for instrument calibration, identification of unknowns, and method validation. | Ketone Mix (C4-C9) [33], 4-Heptanone (for linearity/LOD) [33] |
| Headspace Vials | Contain the sample during incubation; allow for the creation and sampling of the headspace. | 20 mL screw-top headspace vials [39] [33] |
| Culture Media | For growing microorganisms in clinical/food safety studies. | Tryptic Soy Broth (TSB) [34], Nutrient Broth (NB) [35] |
| SPME Fibers | (Optional) For pre-concentration of VOCs to enhance sensitivity. | PDMS/DVB fiber [36] |
| Chromatography Column | The heart of the GC separation. | MXT-WAX (polar) or MXT-5 (non-polar) capillary column [38] [34] |
| High-Purity Gases | Serves as the carrier and drift gas. | Nitrogen (N₂), purity ≥ 99.999% [38] [34] |
GC-IMS has firmly established itself as a robust and highly effective platform for rapid, on-site screening. Its performance profile—characterized by high sensitivity, minimal sample preparation, portability, and rapid analysis times—makes it uniquely suited for applications in food safety and clinical diagnostics where speed and non-destructiveness are critical. While it may not replace the definitive identification power of laboratory-based GC-MS, it serves as a powerful complementary tool that can provide actionable intelligence in real-time. The experimental data confirms its ability to detect early mold contamination in grains and to discriminate between diseased and healthy states in clinical samples based on VOC profiles. As technology advances and reference libraries expand, GC-IMS is poised to play an increasingly vital role in the move towards decentralized, rapid diagnostics and quality control.
The rapid identification and profiling of illicit substances present a significant challenge for law enforcement and forensic laboratories worldwide. Traditional analytical methods, while highly accurate, often require complex sample preparation, lengthy analysis times, and laboratory-bound instrumentation, delaying critical decision-making. Within this context, Gas Chromatography-Ion Mobility Spectrometry (GC-IMS) emerges as a powerful analytical technique that combines the high separation power of gas chromatography with the rapid detection capabilities of ion mobility spectrometry. This case study objectively compares the performance of GC-IMS against established laboratory-based methods for the analysis of seized drug samples, providing experimental data and protocols to illustrate its utility for rapid on-site screening. The research is framed within a broader thesis investigating the viability of GC-IMS as a complementary technology to conventional techniques, particularly in scenarios where speed, portability, and sensitivity are paramount.
GC-IMS is an analytical technique designed for the sensitive detection and separation of volatile organic compounds (VOCs). Its operation involves a two-step process: first, sample volatiles are separated in a capillary column based on their partitioning between a stationary and a mobile phase; second, the separated compounds are ionized and introduced into a drift tube where they are separated based on their size, shape, and charge under the influence of an electric field [1].
The strengths of GC-IMS are particularly well-suited for on-site applications. It enables rapid detection with high sensitivity, allowing for the identification of VOCs in a short time, which is ideal for rapid screening [1]. The technique is user-friendly, typically requiring minimal sample preparation and enabling direct analysis of headspace samples, thereby streamlining the workflow [1]. Furthermore, GC-IMS equipment is often portable and has low maintenance costs compared to traditional laboratory mass spectrometers, offering significant potential for widespread field deployment [1]. A key analytical advantage is its dual-separation power, which provides two data dimensions—retention time and reduced ion mobility—that minimize cross-sensitivities and enhance the reliability of detection [12].
Comparative Performance Data: GC-IMS vs. Established Methods
To objectively evaluate GC-IMS, its performance is compared against two gold-standard laboratory techniques: Gas Chromatography-Mass Spectrometry (GC-MS) and Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS). The following tables summarize key quantitative metrics and application-focused performance indicators.
Table 1: Quantitative Performance Metrics for Drug Analysis
| Performance Parameter | GC-IMS | GC-MS | LC-MS/MS |
|---|---|---|---|
| Detection Limit | Single-digit ppbv (parts per billion by volume) for precursors [12] | Low ppt-ppb range | Low pg-mg levels (in biofluids) |
| Analysis Time | Minutes (Fast response) [1] | 30+ minutes [40] | 30+ minutes (including sample prep) |
| Sample Preparation | Minimal (e.g., centrifugation for oral fluids) [41] | Often requires extraction/derivatization | Requires complex preparation (e.g., SPE) [41] |
| Instrument Portability | Yes [1] | No (Lab-bound) | No (Lab-bound) |
| Qualitative Data | Retention time & reduced ion mobility (K₀) [12] | Retention time & mass spectrum | Retention time & mass fragments |
Table 2: Application-Based Performance Comparison
| Application Scenario | GC-IMS Performance | GC-MS/LC-MS/MS Performance | Supporting Data |
|---|---|---|---|
| On-site Drug Detection | Direct, rapid analysis of oral fluids with <8.4% error vs. reference [41] | Lab-based; sample transport delays | Detected amphetamine, cocaine, ketamine, etc. [41] |
| Clandestine Lab Detection | Detects synthesis precursors & pathway-specific byproducts in headspace [12] | Excellent for definitive profiling but not for on-site air monitoring | Identified BMK synthesis pathways (Dakin-West, nitrostyrene) [12] |
| Throughput & Operational Cost | High throughput, low maintenance costs [1] | High operational cost, requires skilled personnel [42] | LC-MS/MS service contracts: $5k-$20k/year [42] |
Experimental Protocols for Illicit Drug Analysis via GC-IMS
Protocol 1: Detection of Synthesis Precursors in Clandestine Laboratories
This protocol is adapted from a study focused on revealing illicit drug laboratories by detecting relevant chemicals of methamphetamine synthesis [12].
- Objective: To identify volatile precursors and pathway-specific byproducts from the synthesis of benzyl methyl ketone (BMK), a key methamphetamine precursor, in the gas phase.
- Sample Preparation: No complex preparation is needed. A real seized sample of BMK is placed in a headspace vial, and its gaseous headspace is directly sampled [12].
- GC-IMS Conditions:
- Column: FS-SE-54-CB-1 (15 m x 0.53 mm ID).
- Analysis Time: 20 minutes.
- Column Temperature: 60°C.
- Carrier/Drift Gas: Nitrogen.
- IMS Temperature: 45°C.
- Injection Volume: 500 μL (headspace).
- Incubation: 15 minutes at 60°C with 500 rpm agitation [12].
- Data Analysis: Compounds are identified based on their retention time and reduced ion mobility (K₀) using built-in NIST and IMS databases. The presence of specific byproducts allows for the determination of the synthesis pathway (e.g., Dakin-West, nitrostyrene) [12].
- Key Findings: The method successfully detected precursors from three main BMK synthesis pathways at single-digit ppbv levels, demonstrating the potential for profiling suspicious premises by sampling the ambient air [12].
Protocol 2: Determination of Illicit Substances in Oral Fluids
This protocol is based on research developing a GC-IMS method for the analysis of illicit and psychoactive substances in oral fluids, an alternative biological matrix [41].
- Objective: To reliably determine the presence of target drugs like amphetamine, cocaine, and ketamine in oral fluid samples.
- Sample Preparation: A non-invasive oral fluid sample is collected. A simple centrifugation step is sufficient prior to injection, eliminating the need for solid-phase extraction or derivatization [41].
- GC-IMS Conditions: (General parameters as in Protocol 4.1, with method-specific tuning for the target analytes).
- Data Analysis: Identification is performed using a published database of reduced ion mobility constants (K₀) for approximately 200 psychoactive compounds. For unequivocal identification, verification with MS is recommended, though the K₀ database provides robust preliminary information [41].
- Key Findings: When analyzing certified reference materials, the method demonstrated high reliability, with relative percentage errors lower than 8.4% for the target substances, confirming its suitability for sensitive and selective on-site screening [41].
Visualizing the GC-IMS Workflow for Seized Drug Analysis
The following diagram illustrates the logical workflow and analytical process for analyzing seized drug samples using GC-IMS, from sample collection to final reporting.
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details key reagents, materials, and instrumentation essential for conducting GC-IMS experiments in the context of illicit drug analysis.
Table 3: Essential Research Reagent Solutions for GC-IMS Drug Analysis
| Item | Function/Description | Application in Protocol |
|---|---|---|
| GC-IMS Instrument | Portable or benchtop system (e.g., FlavorSpec) with a drift tube filled with nitrogen drift gas. | Core analytical instrument for all separation and detection [12]. |
| Headspace Vials & Septa | Sealed vials for containing solid/liquid samples and allowing volatile accumulation in the headspace. | Essential for headspace sampling of seized materials (Protocol 4.1) [12]. |
| Nitrogen Gas (High Purity) | Serves as both the carrier gas for GC and the drift gas for IMS. | Critical for maintaining instrument function and achieving separation [12]. |
| Certified Reference Materials | Authentic, pure standard solutions of target drugs and precursors (e.g., amphetamine, BMK). | Used for instrument calibration, method development, and validation [41]. |
| IMS Database (K₀ Values) | A library of reduced ion mobility constants for psychoactive substances. | Enables preliminary identification of unknowns without MS confirmation [41]. |
| Oral Fluid Collection Kit | Non-invasive devices for collecting oral fluid samples from subjects. | Required for sample collection in roadside or clinical testing (Protocol 4.2) [41]. |
| Centrifuge | Laboratory equipment for separating solids from liquids in a sample. | Used for the simple preparation of oral fluid samples (Protocol 4.2) [41]. |
This case study demonstrates that GC-IMS is a compelling technology for the rapid analysis of seized drugs and illicit substance profiling. Its key advantages of high sensitivity (single-digit ppbv), rapid analysis, minimal sample preparation, and instrument portability position it as a powerful tool for on-site screening. While established lab-based methods like GC-MS and LC-MS/MS remain the gold standard for definitive confirmation and non-volatile metabolite analysis [20], GC-IMS effectively bridges the gap between the need for speed in the field and analytical rigor. By providing reliable detection of synthesis precursors, pathway-specific byproducts, and drugs in biological fluids like oral fluid, GC-IMS empowers law enforcement and forensic professionals with timely intelligence, ultimately supporting faster interventions and strengthening public safety efforts.
Navigating Challenges: Matrix Effects, Data Complexity, and Performance Optimization
For researchers pursuing rapid, on-site analysis of volatile organic compounds (VOCs), Gas Chromatography coupled with Ion Mobility Spectrometry (GC-IMS) presents a powerful alternative to traditional lab-bound methods like GC-MS. However, the transition from controlled laboratory environments to the field introduces significant analytical challenges, primarily from matrix effects. Among these, variable humidity and complex sample backgrounds are critical factors that can profoundly influence ionization efficiency and signal stability, potentially compromising quantification accuracy and detection reliability. This guide objectively compares the performance of GC-IMS with GC-MS in managing these matrix effects, providing a detailed examination of their ionization mechanisms, supporting experimental data, and standardized protocols to empower robust method development.
Ionization Fundamentals and Matrix Vulnerability
The core difference in how GC-IMS and GC-MS handle matrix effects stems from their fundamental ionization processes.
GC-IMS Ionization at Atmospheric Pressure
The ionization process in a typical GC-IMS, often employing a radioactive β-emitter (³H or ⁶³Ni), occurs at atmospheric pressure and is governed by gas-phase chemical reactions [43]. Beta particles ionize the drift gas (often nitrogen or clean air), leading to a cascade of reactions that produce stable reactant ions (RIP), typically protonated water clusters (H₃O⁺(H₂O)ₙ) [3] [43]. When analyte molecules (M) elute from the GC column, they interact with these reactant ions. Depending on the analyte's proton affinity and concentration, this results in the formation of protonated monomers (MH⁺(H₂O)ₙ₋ₓ) or proton-bound dimers (M₂H⁺(H₂O)ₘ₋ₓ) [43]. The separation that follows is based on the ion's mobility in a drift tube, which is a function of its mass, charge, and collision cross-section (a measure of its size and shape) [3] [44].
GC-MS Ionization under Vacuum
In contrast, GC-MS typically relies on electron impact (EI) ionization, which occurs under high vacuum. In this process, sample molecules are bombarded with high-energy electrons (usually 70 eV), leading to the ejection of an electron and the formation of a molecular ion (M⁺•) that often undergoes extensive fragmentation [45]. This vacuum-based process is largely isolated from the influence of ambient conditions like humidity.
The following diagram illustrates the distinct ionization pathways in GC-IMS and how they are susceptible to interference from humidity and sample background, in contrast to the more isolated GC-MS process.
Diagram: Ionization pathways contrasting GC-MS (vacuum) and GC-IMS (atmospheric). The GC-IMS process is vulnerable to interference from humidity and sample matrix, leading to signal changes.
Comparative Performance Data in Controlled Studies
The theoretical vulnerabilities of GC-IMS translate into measurable, compound-specific effects. Experimental data reveals how humidity and sample complexity impact quantification.
Quantifying the Impact of Humidity
A systematic study investigating the effect of carrier gas humidity on positive mode IMS provides crucial quantitative insights [46]. The research demonstrated that while the drift times (and thus peak positions) remained constant, signal intensities were significantly affected for many compound classes. The degree of impact, however, varied substantially.
Table 1: Impact of Humid Carrier Gas on IMS Signal Intensity [46]
| Compound Class | Representative Compounds | Signal Intensity Trend | Magnitude of Change |
|---|---|---|---|
| Amines | Various amines | Comparatively unaffected | Constant over humidity up to 2000 ppmv H₂O |
| Toluenes | Toluene, Xylene | Significant decrease | Strong reduction with increasing humidity |
| Chlorinated Compounds | Chlorobenzenes | Significant decrease | Strong reduction with increasing humidity |
| Ketones | Acetone, Butanone | Varies by specific ion | Different product ions show different behaviors |
Sensitivity and Dynamic Range: GC-IMS vs. GC-MS
A comprehensive assessment of a TD-GC-MS-IMS system provided a direct comparison of the two detectors' performance under identical conditions [18]. This study highlighted a classic trade-off between supreme sensitivity and broad dynamic range.
Table 2: Quantitative Performance Comparison of GC-IMS and GC-MS [18]
| Performance Metric | GC-IMS | GC-MS |
|---|---|---|
| Relative Sensitivity | ~10x more sensitive for ketones | Baseline (1x) |
| Typical Limit of Detection | Picogram per tube range | Higher than IMS |
| Linear Dynamic Range | ~1 order of magnitude (e.g., 0.1–1 ng/tube for pentanal) | ~3 orders of magnitude (up to 1000 ng/tube) |
| Response at High Concentrations | Transitions to logarithmic response | Maintains linearity |
| Key Quantification Advantage | Superior for trace-level detection | Reliable across a wide concentration range |
Experimental Protocols for Mitigating Matrix Effects
Robust experimental design is essential for managing matrix effects. The following protocols, drawn from research, provide a framework for reliable analysis.
Protocol for Evaluating Humidity Effects
Objective: To characterize the impact of specific humidity levels on the signal intensity of target analytes.
- Standard Generation: Utilize a dynamic dilution system for a test mixture of relevant compounds (e.g., anilines, toluenes, chlorobenzenes, ketones) [46].
- Humidity Control: Employ a gas supply with a mass flow controller. Split the flow, directing one stream through a water-filled bubbler and the other remaining dry. Precisely mix the streams using mass flow controllers to achieve a defined relative humidity (e.g., from 0% to 80% at 30°C) [46].
- Data Acquisition: Introduce the humidified standard into the GC-IMS. Acquire data for all compounds across the defined humidity range.
- Data Analysis: For each compound, plot the normalized signal intensity against the absolute water concentration (e.g., ppmv). Determine the humidity susceptibility factor for each analyte [46].
Protocol for Long-Term Stability Assessment
Objective: To determine the reproducibility of retention time, drift time, and signal intensity over an extended period, critical for validating methods against day-to-day environmental fluctuations.
- System Calibration: Use a certified calibration mixture of ketones or other stable VOCs [18].
- Data Collection: Perform repeated measurements (e.g., on 156 measurement days over 16 months) using identical protocols [18].
- Precision Calculation: Calculate the Relative Standard Deviations (RSDs) for:
Data Processing Workflow for Non-Targeted Screening
For complex samples, advanced data processing is key to extracting meaningful information despite background noise and signal drift.
- Pre-processing: Load raw data and apply denoising, baseline correction, and spectral alignment to correct for instrumental variances [3] [47].
- Feature Extraction: Perform automatic peak detection and integration across the 2D (retention time vs. drift time) landscape [3] [47].
- Peak Clustering: Group corresponding peaks across all samples to account for minor misalignments [3].
- Multivariate Analysis: Use the final peak table for statistical analysis (e.g., PLS-DA, PCA) to identify features most relevant for class separation, effectively filtering out irrelevant background [3] [47].
The Scientist's Toolkit: Key Reagents and Materials
Successful implementation of GC-IMS methods, particularly for managing matrix effects, relies on specific consumables and reagents.
Table 3: Essential Research Reagent Solutions for GC-IMS Analysis
| Item | Function & Importance | Justification |
|---|---|---|
| Thermal Desorption (TD) Tubes | Sample collection and pre-concentration of VOCs from air/gas matrices; essential for trace-level analysis. | Enables standardized sampling beyond the lab; critical for reproducibility in clinical/environmental studies [18]. |
| Sorbent Material (e.g., Tenax TA) | Packing material inside TD tubes; determines the range of volatilities and compound classes that can be trapped. | Choice of sorbent defines the analytical scope, from high volatiles to semi-volatiles [18]. |
| High-Purity Drift Gas | Nitrogen or clean, dry air used as the counter-gas in the IMS drift tube. | Purity is critical for stable reactant ion populations and reproducible drift times [4] [43]. A moisture trap is often recommended [4]. |
| Certified Calibration Solutions | Mixtures of target analytes in suitable solvents (e.g., methanol) for creating external calibration curves. | Required for quantification. Preparation of stock solutions (e.g., for aldehydes, ketones, alcohols) must be controlled for accuracy [18]. |
| Internal Standards (IS) | Deuterated or otherwise isotopically labeled analogs of target analytes. | When applicable, an IS can correct for losses during sample preparation and fluctuations in ionization efficiency, improving quantification accuracy [18]. |
GC-IMS stands out as a highly sensitive, portable technology ideal for rapid on-site screening where detecting trace-level VOCs is paramount. However, its analytical performance is inherently linked to the sample matrix. As the data shows, humidity can suppress signals for key compound classes, and co-eluting analytes can compete for charge, leading to non-linear responses. GC-MS, while less portable and often less sensitive, provides a more robust platform for quantifying analytes across a wide dynamic range due to its stable, vacuum-based ionization.
The choice between these techniques is not a matter of superiority but of strategic application. For on-site food freshness screening, environmental monitoring, or clinical breath analysis, GC-IMS is a powerful "Swiss army knife" [16], provided that methods are rigorously calibrated against matrix effects using the protocols and reagents outlined. For absolute quantification of diverse analytes in unknown matrices, lab-based GC-MS remains the reference standard. By understanding and actively managing these ionization vulnerabilities, researchers can fully leverage the speed and sensitivity of GC-IMS for confident, on-the-spot analysis.
Gas Chromatography-Ion Mobility Spectrometry (GC-IMS) represents a powerful analytical technique that combines the separation power of gas chromatography with the high sensitivity and rapid detection capabilities of ion mobility spectrometry [1]. This hybrid technology has gained significant traction in various fields, including food authentication, clinical diagnostics, and environmental monitoring, due to its ability to provide rapid, sensitive detection of volatile organic compounds (VOCs) with minimal sample preparation [16]. The fundamental strength of GC-IMS lies in its two-dimensional separation: compounds are first separated by their retention time in the GC column and subsequently by their drift time in the IMS drift tube [3].
However, the data generated by GC-IMS presents unique analytical challenges. The raw data is highly dimensional, complex, and often suffers from technical artifacts including strong non-linearities, baseline problems, misalignments, peak overlaps, and long peak tails [3]. These challenges must be systematically addressed through comprehensive data processing workflows to extract meaningful chemical information from samples. The data analysis pipeline typically progresses through three critical stages: data pre-processing, peak picking (feature extraction), and finally, statistical analysis and interpretation [48]. This guide demystifies each stage of GC-IMS data analysis, providing researchers with practical frameworks for handling the complexities of this powerful analytical technique.
GC-IMS in the Analytical Landscape: A Performance Comparison
Understanding the position of GC-IMS within the broader analytical toolkit requires a clear comparison with established laboratory techniques, particularly GC-MS. The table below summarizes the key performance characteristics and operational considerations.
Table 1: Comparison of GC-IMS with GC-MS for VOC Analysis
| Parameter | GC-IMS | GC-MS |
|---|---|---|
| Sensitivity | ~10x more sensitive than MS in a TD-GC-MS-IMS system; LOD in picogram/tube range [18] | High sensitivity, but lower than IMS in coupled systems [18] |
| Linear Dynamic Range | ~1 order of magnitude (can be extended to 2 orders with linearization) [18] | ~3 orders of magnitude (up to 1000 ng/tube) [18] |
| Analysis Speed | Rapid analysis; suitable for on-site, high-throughput screening [1] | Typically slower, laboratory-based method |
| Portability & Footprint | Portable systems available; smaller footprint, no vacuum system required [3] [16] | Generally non-portable, larger footprint, requires sophisticated vacuum [3] |
| Operational Costs & Greenness | Lower energy consumption; can use air as carrier gas; aligns with Green Analytical Chemistry principles [16] | Higher energy consumption; often uses helium (non-renewable); greater resource intensity [16] |
| Data Complexity | High-dimensional 2D data (retention time vs. drift time); requires specialized pre-processing [3] | 2D data (retention time vs. m/z); well-established data analysis libraries |
| Compound Identification | Lacks universal database; identification often requires parallel MS or internal standards [18] | Powerful identification via extensive, universal mass spectral libraries [18] |
This comparison highlights that GC-IMS excels in applications requiring high sensitivity, rapid results, and portability for on-site analysis, while GC-MS remains the standard for applications demanding broad linear dynamic range and confident identification of unknown compounds across a wide concentration range.
The Data Analysis Workflow: From Raw Data to Chemical Information
The journey from raw GC-IMS signals to actionable results requires a structured, multi-stage workflow. This process mitigates the inherent technical challenges of the data and extracts robust features for subsequent statistical modeling.
Data Pre-processing: Correcting Technical Variability
The first and crucial step is data pre-processing, which aims to correct for instrumental artifacts and technical variations to ensure comparability between samples. Key pre-processing steps include:
- Baseline Correction: The signal baseline in GC-IMS can fluctuate due to detector noise and background drift. Effective baseline correction is essential for accurate peak detection and integration [3].
- Denoising and Smoothing: Digital smoothing techniques are applied to reduce high-frequency noise, which improves the signal-to-noise ratio without distorting the underlying chemical information [3].
- Alignment (Retention Time and Drift Time): Misalignments in both the chromatographic retention time and ion mobility drift time axes are a major challenge. These can arise from minor fluctuations in carrier gas flow, temperature, or drift gas conditions. Sophisticated 2D alignment algorithms are necessary to ensure that the same feature across different samples is correctly matched [3] [48]. This step is critical for any comparative study.
- RIP Details (Optional): In some workflows, the Reactant Ion Peak (RIP), which represents the charge reservoir (e.g., H₃O⁺(H₂O)ₙ clusters), may require specific processing, such as detailing or normalization [3].
Peak Picking and Feature Extraction
After pre-processing, the next step is "peak picking" or feature extraction, where the relevant chemical signatures are identified and quantified from the complex 2D landscape. Given the high dimensionality of raw data, this step is vital for reducing data complexity before statistical analysis [3]. There are several strategic approaches:
- Manual Feature Selection (Targeted Analysis): When analytes of interest are known a priori, researchers can use vendor or custom software to manually select and integrate specific peaks, often building an in-house database [3]. This is common for quality control or when tracking specific biomarkers.
- Automated Peak Picking (Untargeted Analysis): For untargeted fingerprinting analyses, automated algorithms detect potential features (peaks) across the entire 2D data matrix. These algorithms typically identify local maxima in the signal and define the boundaries of each peak [3].
- Feature Clustering: Due to small tolerances in peak locations across samples, clustering methods are often employed post-detection to group features believed to originate from the same chemical compound, creating a consolidated list of variables for all samples [3].
- Alternative Data Reduction Strategies: Other effective strategies include extracting the total area of the reactant ion peak chromatogram (RIC) or using the full RIC response, which collapses the 2D data into a 1D chromatogram, simplifying subsequent analysis [3].
The choice of strategy is often a trade-off between the amount of chemical information preserved and the computational effort required [3].
Dimensionality Reduction and Statistical Modeling
Once a feature table (samples vs. peak volumes) is generated, chemometric techniques are used to find patterns and build predictive models.
- Dimensionality Reduction: Techniques like Principal Component Analysis (PCA) are used to visualize sample groupings and identify the most influential features (volatile compounds) driving the differences between sample classes [37].
- Classification and Regression: Supervised methods like Partial Least Squares-Discriminant Analysis (PLS-DA), Linear Discriminant Analysis (LDA), or Support Vector Machines (SVM) can be applied to classify samples based on their GC-IMS fingerprints or to quantify specific properties [3].
Diagram: GC-IMS Data Analysis Workflow
Experimental Protocols for Robust GC-IMS Analysis
Long-Term Stability and Precision Assessment
A comprehensive study assessed the long-term stability of a TD-GC-IMS system over 16 months (156 measurement days) using ketones as reference compounds. The methodology and key findings provide a benchmark for system performance [18].
- Methodology: A mobile flow- and temperature-controlled sampling unit for thermal desorption (TD) tubes was developed to ensure standardized application. This allowed for the introduction of both gaseous and liquid samples with high reproducibility.
- Quantitative Results: The system demonstrated high stability, with relative standard deviations (RSDs) for signal intensities ranging from 3% to 13%. The retention time deviations were very low, between 0.10% and 0.22%, and drift time deviations ranged from 0.49% to 0.51% [18].
- Significance: This level of long-term precision confirms that GC-IMS is a robust technique suitable for routine VOC analysis and long-term monitoring studies, where reproducibility is critical.
Improving Quantification: Linearization of IMS Response
A key limitation of IMS is its narrow linear dynamic range compared to MS. A recent study addressed this challenge with a specific methodological approach [18].
- Methodology: Researchers evaluated the quantification performance of IMS versus MS by analyzing a series of aldehydes and ketones. The IMS response for compounds like pentanal was found to be linear over only one order of magnitude (e.g., 0.1 to 1 ng/tube) before transitioning into a logarithmic response.
- Intervention: To improve IMS quantification, the study implemented a linearization strategy. This mathematical processing of the signal extended the usable calibration range from one to two orders of magnitude [18].
- Significance: This approach enhances the utility of IMS for quantitative analysis, making it more competitive with traditional detectors for applications requiring quantification over broader concentration ranges.
Essential Research Reagent Solutions
Successful GC-IMS analysis relies on a set of key reagents and materials for calibration, sampling, and system maintenance. The following table details these essential components.
Table 2: Key Research Reagents and Materials for GC-IMS
| Item | Function & Purpose | Example Application / Note |
|---|---|---|
| Thermal Desorption (TD) Tubes | Capture and concentrate VOCs from air or headspace; filled with specific adsorbent materials. | Enables sampling beyond the laboratory; requires controlled flow/temp for reproducible adsorption of liquid standards [18]. |
| Chemical Standards | High-purity (>95%) volatile compounds for system calibration and identification. | Used to prepare calibration solutions (e.g., alcohol, aldehyde, ketone groups in methanol) [18]. |
| n-Alkane Series (e.g., C7-C40) | Used for calculating Linear Retention Indices (LRI or I_T) to normalize retention times. | Critical for compensating for run-to-run retention time shifts and aiding in compound identification [49]. |
| Inert Carrier Gas | Mobile phase for GC separation; also serves as drift gas in IMS. | Nitrogen or air can be used, reducing reliance on helium and operational costs [16]. |
| Derivatization Reagents (e.g., MTBSTFA) | Chemically modify non-volatile analytes (e.g., metabolites) to make them volatile for GC analysis. | Used in metabolomics studies of biological fluids like plasma [49]. |
GC-IMS has firmly established itself as a powerful and versatile "Swiss army knife" for volatile compound analysis [16]. Its strengths in sensitivity, speed, and portability make it exceptionally well-suited for rapid on-site screening and high-throughput applications. While the data it generates present unique challenges in the form of high dimensionality and technical artifacts, established and evolving data analysis workflows—encompassing robust pre-processing, strategic feature extraction, and advanced chemometrics—effectively demystify this complexity. By understanding and implementing these protocols, researchers can leverage the full potential of GC-IMS as a greener, faster, and highly sensitive complement to traditional laboratory-based methods like GC-MS.
Gas Chromatography-Ion Mobility Spectrometry (GC-IMS) represents a powerful analytical technique that combines the superior separation capabilities of gas chromatography with the rapid detection and high sensitivity of ion mobility spectrometry. This hybrid technology is particularly valuable for the analysis of volatile organic compounds (VOCs) across diverse fields including food authenticity testing, medical diagnostics, fragrance analysis, and environmental monitoring [1] [50]. The analytical performance of GC-IMS—encompassing sensitivity, resolution, and analysis speed—is profoundly influenced by three fundamental instrumental parameters: carrier/drift gas flows, temperature regimes, and injection volume. Proper optimization of these parameters is especially critical for applications requiring rapid on-site screening, where the balance between analytical performance and operational practicality must be carefully managed [51] [52].
The fundamental operating principle of GC-IMS involves the initial separation of volatile compounds in a chromatographic column, followed by ionization and separation in the drift tube based on ion mobility in an electric field [14] [4]. This two-dimensional separation process provides orthogonality, with GC separation primarily based on compound volatility and stationary phase interactions, while IMS separation depends on the ion's mass, charge, and collision cross-section in the gas phase [4]. Understanding this foundational principle is essential for meaningful parameter optimization, as each dimension of separation responds differently to instrumental adjustments.
The Critical Role of Gas Flow Dynamics
Fundamental Flow Principles in GC-IMS
The gas flow dynamics within a GC-IMS system represent a complex interplay between the carrier gas transporting analytes through the GC column and the drift gas responsible for moving ions through the drift tube. Successful GC analysis requires careful control of carrier gas, as it directly impacts retention times, retention factors, and all subsequent calculations based on these parameters [53]. The average linear velocity of the carrier gas, typically expressed in cm/s, plays a crucial role in determining separation efficiency and analysis time. Optimal flow rates must strike a balance between providing sufficient separation in the GC dimension and ensuring compatibility with the IMS detector requirements [53].
A key challenge in GC-IMS coupling involves managing the significant discrepancy between the relatively low flow rates through capillary GC columns (typically 1-2 mL/min) and the substantially higher flow rates required through IMS drift tubes [4]. This flow imbalance can lead to peak broadening if not properly addressed through instrumental design or the use of makeup gas. The fundamental relationship between column dimensions and IMS internal volume dictates that the ratio of the internal volume of the IMS reaction region to the internal volume of the GC column is a critical factor determining overall system efficiency [4].
Practical Flow Optimization Strategies
Table 1: Gas Flow Configurations and Their Analytical Implications in GC-IMS
| Flow Parameter | Typical Range | Impact on Separation | Considerations for On-site Applications |
|---|---|---|---|
| Carrier Gas Flow Rate | 1-3 mL/min (capillary columns) | Higher flows reduce retention times but may compromise GC resolution; lower flows improve separation but extend analysis time | Nitrogen preferred for portability and cost; helium provides better efficiency but less suitable for field deployment |
| Drift Gas Flow Rate | 100-500 mL/min | Higher flows reduce drift times and may improve peak shape by minimizing ion-molecule interactions | Clean, dry air can be used instead of high-purity nitrogen for cost-effective field operation |
| Makeup Gas Flow | Often required for capillary-to-IMS interface | Prevents peak broadening by compensating for flow rate disparity between GC and IMS | Adds system complexity and gas consumption, requiring careful design for portable instruments |
| Flow Control Mode | Constant flow vs. constant pressure | Constant flow provides more consistent retention times in temperature-programmed runs | Constant flow mode preferred for method transferability between instruments |
Modern GC-IMS systems employ various strategies to optimize the flow dynamics between the separation and detection components. One common approach involves the use of larger diameter GC columns (320 μm or 530 μm) or multi-capillary columns (MCC) that provide higher internal volume while maintaining reasonable resistance to mass transfer [4]. Alternatively, specialized drift tube designs with minimized internal volume and optimized gas flow architectures have been developed to reduce the required makeup gas flows [51] [4]. These optimized flow configurations create laminar flow profiles that guide the sample directly to the gas outlet, effectively reducing diffusion within the reaction region and improving chromatographic peak shape [51].
For on-site applications, practical considerations often favor the use of nitrogen as both carrier and drift gas, as it eliminates the need for multiple gas cylinders and reduces operational costs [51] [4]. The implementation of effective moisture traps in the drift gas line is particularly important for field applications, as humidity fluctuations can significantly impact ionization efficiency and mobility measurements [4].
Temperature Optimization Strategies
Temperature Effects on Separation and Detection
Temperature represents one of the most critical parameters for optimizing GC-IMS performance, influencing both the chromatographic separation and ion mobility detection processes. In the GC dimension, temperature directly affects partitioning between stationary and mobile phases, with higher temperatures generally reducing retention times but potentially compromising separation efficiency [53]. In the IMS dimension, temperature impacts ion mobility through its effects on ion-neutral collision cross-sections and cluster formation processes [52]. The relationship between ion mobility (K) and temperature is described by the Mason-Schamp equation, where K is proportional to the absolute temperature, assuming the ion-neutral cross-section term remains nearly temperature-independent [52].
A significant challenge in conventional GC-IMS systems has been the limited temperature range of commercially available drift tubes, typically restricted to 45-90°C [52]. This limitation becomes particularly problematic when analyzing higher-boiling compounds such as terpenes and phenylpropanoids, which are common in fragrance, cosmetic, and natural product applications. At these lower temperatures, high-boiling compounds often experience significant peak tailing due to condensation or adsorption effects within the IMS cell [51] [52]. This tailing effect can severely compromise resolution, particularly when co-elution of different compounds occurs in complex matrices.
High-Temperature IMS Solutions
Table 2: Temperature Optimization Effects on Different Compound Classes in GC-IMS
| Compound Class | Optimal Drift Tube Temperature | Observed Effect | Practical Application |
|---|---|---|---|
| Monoterpenes | 120-180°C | Significant reduction in peak tailing; improved resolution of structural isomers | Analysis of essential oils, fragrance allergens in cosmetics |
| Phenylpropanoids | 120°C | Resolution of critical pairs (eugenol/isoeugenol) above 1.5 | Quality control of natural products, spice authenticity |
| Ketones | 140-160°C | Optimal aspect ratio achieved in intermediate temperature range | Food flavor analysis, fermentation monitoring |
| Alcohols | 120-140°C | Improved peak symmetry at moderate temperature increase | Beverage analysis, fuel oxygenates detection |
| Aldehydes | Progressive improvement up to 180°C | Steady improvement in peak shape with increasing temperature | Lipid oxidation monitoring, indoor air quality |
Recent advancements in IMS instrumentation have addressed these limitations through the development of high-temperature IMS (HTIMS) systems capable of operating at drift tube temperatures up to 180°C [51] [52]. Research has demonstrated that increasing the IMS drift tube temperature significantly improves peak shapes for high-boiling compounds, with one study reporting substantial reduction in peak tailing for monoterpenes like geraniol, carvone, pinene, citral, and β-caryophyllene [51] [52]. This improvement is attributed to the reduction of condensation and adsorption effects within the IMS cell at elevated temperatures.
The temperature optimization strategy must consider the specific compound classes being analyzed. For instance, while most monoterpenes and phenylpropanoids show progressive improvement in peak shape up to 180°C, ketones and alcohols often exhibit optimal peak symmetry at intermediate temperatures (140-160°C and 120-140°C, respectively) [52]. This substance-dependent temperature response necessitates method-specific optimization, particularly for targeted analyses. Furthermore, temperature programming in the GC dimension must be coordinated with IMS temperature settings to ensure compatibility across the entire analytical workflow.
For on-site screening applications, the energy consumption associated with elevated IMS temperatures must be balanced against analytical requirements. Portable systems may implement power-saving strategies such as standby modes with reduced temperature settings, with rapid heating to operational temperatures when analysis is initiated.
Injection Volume Considerations
Injection Volume Effects on Ionization and Detection
Injection volume represents a crucial parameter in GC-IMS method development, directly influencing sensitivity, peak shape, and the formation of ion species. In IMS detection, the analyte concentration significantly affects the formation of monomers and dimers, with higher concentrations promoting the formation of proton-bound dimers (M₂H⁺) in addition to protonated monomers (MH⁺) [52]. This concentration-dependent ionization behavior can complicate spectral interpretation if not properly controlled through optimized injection parameters.
The relationship between injection volume and analyte concentration is particularly important given the narrow linear dynamic range of IMS detection, which typically spans from approximately 1 ppb to 1 ppm depending on the analyte [14]. Outside this range, quantitative analysis becomes challenging due to the non-linear response resulting from competitive ionization processes and the formation of different ion species at varying concentrations [14]. This limitation necessitates careful optimization of injection volume to ensure target analytes fall within the instrument's linear response range.
Injection Volume Optimization Approaches
Table 3: Comparison of Sample Introduction Methods in GC-IMS
| Introduction Method | Typical Volume | Advantages | Limitations | Suitable Applications |
|---|---|---|---|---|
| Static Headspace | 100-1000 µL | Minimal sample preparation; avoids non-volatile matrix components | Limited sensitivity for less volatile analytes | Food freshness monitoring, polymer quality control |
| Headspace-SPME | N/A (extraction) | Pre-concentration capability; improved sensitivity | Longer sample preparation time; fiber degradation | Trace analysis in complex matrices (e.g., forensic, environmental) |
| Direct Vapor Injection | 10-100 µL | Simple implementation; rapid analysis | Potential for contamination; limited sensitivity | Process monitoring, breath analysis |
| Thermal Desorption | Variable | High sensitivity; suitable for low-volume air samples | Requires specialized equipment; potential for artifact formation | Indoor air quality, material emissions testing |
Practical injection volume optimization must consider the entire sample introduction workflow, including headspace equilibration conditions, sample concentration, and the specific detection limits required for target analytes. For liquid samples, common injection volumes range from 100-500 µL for headspace analysis, while gas samples may involve larger volumes to achieve adequate sensitivity [53]. The use of pre-concentration techniques such as solid-phase microextraction (SPME) or thermal desorption can effectively increase the effective injection volume without overwhelming the analytical system [50].
The optimal injection volume represents a compromise between sufficient sensitivity for trace-level analytes and avoiding detector overloading or significant changes in ionization behavior. Method development typically involves injecting a series of standard solutions at different concentrations or volumes to establish the linear dynamic range for each target compound. For untargeted screening applications, a moderate injection volume that provides adequate sensitivity for most expected compounds without significant peak broadening or distortion is preferred.
For rapid on-site screening, simplified sample introduction approaches such as direct vapor injection or static headspace sampling are often employed to minimize sample preparation time and complexity. These approaches may require slightly larger injection volumes compared to laboratory-based methods to compensate for the absence of pre-concentration steps, though careful optimization remains essential to maintain analytical performance.
Experimental Protocols for Parameter Optimization
Systematic Approach to Flow Rate Optimization
A methodical approach to gas flow optimization begins with establishing baseline separation conditions using manufacturer-recommended settings, followed by systematic variation of individual parameters while monitoring key performance indicators. The following protocol outlines a comprehensive strategy for optimizing carrier and drift gas flows:
Initial System Characterization: Determine the current system performance by analyzing a test mixture containing compounds representative of the target analytes. The test mixture should include at least 5-7 compounds spanning a range of volatilities and functional groups. Record retention times, peak widths, peak symmetry (tailing factors), and signal-to-noise ratios for each compound.
Carrier Gas Flow Rate Optimization: While maintaining constant drift gas flow, systematically vary the carrier gas flow rate across a reasonable range (e.g., 0.5-3.0 mL/min for typical capillary columns). For each flow rate, analyze the test mixture and calculate the height equivalent to a theoretical plate (HETP) for well-resolved peaks. Plot HETP against linear velocity to identify the optimal flow conditions that provide the best compromise between separation efficiency and analysis time.
Drift Gas Flow Rate Optimization: With the carrier gas flow set at the optimized value, systematically vary the drift gas flow rate while analyzing the test mixture. Monitor both the IMS drift times and peak shapes, as increased drift gas flow typically reduces drift times but may also influence peak broadening through changes in ion-neutral collision frequency.
Makeup Gas Optimization: If the system includes provision for makeup gas, optimize this parameter to minimize peak broadening while maintaining adequate sensitivity. The optimal makeup gas flow typically represents 10-30% of the total gas flow entering the IMS detector.
Response Surface Methodology: For critical applications, employ experimental design approaches such as central composite design to model the interactive effects of carrier and drift gas flows on multiple response variables simultaneously.
Temperature Optimization Protocol
Temperature optimization requires careful consideration of both GC column temperature programming and IMS drift tube temperature. The following protocol addresses both dimensions:
GC Temperature Program Optimization: Begin with the GC column temperature program, using isothermal holds or slow temperature ramps to establish baseline separation of critical pairs in the test mixture. Once acceptable GC separation is achieved, proceed to IMS temperature optimization.
IMS Drift Tube Temperature Screening: Conduct initial experiments at a moderate IMS temperature (e.g., 80°C) and analyze the test mixture, paying particular attention to peak shapes for later-eluting, higher-boiling compounds. Systematically increase the IMS temperature in 20°C increments up to the maximum allowable temperature (typically 180°C for HTIMS systems), recording peak parameters at each temperature.
Compound-Specific Temperature Optimization: For each compound in the test mixture, plot peak symmetry (tailing factor) against IMS temperature to identify optimal temperatures for different compound classes. Determine if a single temperature provides acceptable performance for all target analytes or if compromises are necessary.
Mobility Shift Characterization: Record the reduced mobility (K₀) values for each compound at different temperatures. Note that K₀ values typically show temperature dependence, with the magnitude and direction of shift potentially providing structural information [52]. Document these shifts for future compound identification.
Final Method Integration: Once optimal IMS temperature is established, make minor adjustments to the GC temperature program if necessary to fine-tune overall separation. Validate the final method with representative samples to ensure robustness.
Injection Volume Optimization Methodology
The following protocol provides a systematic approach to injection volume optimization:
Linear Range Determination: Prepare a dilution series of standard solutions spanning at least three orders of magnitude in concentration. Using a fixed, moderate injection volume, analyze each standard and plot peak area against concentration to determine the linear dynamic range for each target compound.
Injection Volume Screening: Select a standard concentration near the mid-point of the established linear range. Systematically vary the injection volume (typically spanning 10-500 µL for headspace injections) while analyzing this standard, monitoring both signal response and peak shape.
Ion Species Monitoring: Note the formation of dimer ions at higher injection volumes, as evidenced by the appearance of additional peaks in the ion mobility spectrum with approximately twice the drift time of the monomer peaks. Determine the injection volume threshold at which significant dimer formation begins.
Matrix Effect Evaluation: Repeat the injection volume screening using representative sample matrices rather than pure standards to account for potential matrix effects on ionization efficiency.
Final Volume Selection: Choose an injection volume that provides adequate sensitivity for the least abundant target compounds while remaining within the linear range and avoiding significant dimer formation for the most abundant compounds.
GC-IMS Workflow and Parameter Interrelationships
The optimization of GC-IMS parameters must consider the interconnected nature of the entire analytical workflow. The following diagram illustrates the key parameter interrelationships and their collective impact on analytical performance:
This workflow diagram illustrates the complex interrelationships between instrumental parameters and their collective impact on the final analytical results. The optimization process requires careful consideration of these interactions, as adjustments to one parameter often necessitate compensatory changes to others. For instance, increasing the IMS temperature to improve peak shape for high-boiling compounds may require adjustment of the GC temperature program to maintain optimal elution patterns. Similarly, changes to carrier gas flow rates affect both GC separation efficiency and the transfer of analytes to the IMS detector, potentially influencing ionization dynamics and mobility measurements.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Essential Research Reagents and Materials for GC-IMS Method Development
| Category | Specific Items | Function/Purpose | Application Notes |
|---|---|---|---|
| Calibration Standards | Ketones (C4-C10), alcohols (C3-C8), terpenes, aromatic compounds | System performance verification; retention index calibration; response linearity assessment | Select compounds representing target analyte classes; include n-alkanes for retention index calculation |
| Test Mixtures | Custom blends of volatiles at known concentrations | Method development and optimization; system suitability testing | Should include compounds with varying functional groups and volatilities to assess comprehensive system performance |
| Drift Gases | High-purity nitrogen, dried synthetic air, zero air | Ion transport medium; defines collision environment for mobility separation | Must include appropriate filtration (moisture, hydrocarbons) to minimize background interference |
| Reference Materials | Certified essential oils, fragrance blends, headspace standards | Method validation; quality control; inter-laboratory comparison | Commercially available certified reference materials preferred for validated methods |
| Sample Introduction | Headspace vials, SPME fibers, gas-tight syringes, thermal desorption tubes | Controlled sample introduction with minimal contamination | Material compatibility with target analytes must be verified to avoid adsorption losses |
| System Maintenance | Tritium foil cleaning kits, column connectors, septa, ferrules | Ongoing system performance maintenance | Regular maintenance essential for reproducible performance, especially for portable systems |
The selection of appropriate research reagents and materials is fundamental to successful GC-IMS method development and validation. Calibration standards should encompass a range of compound classes and volatilities to comprehensively characterize system performance across the entire analytical space. For quantitative applications, internal standards (ideally deuterated analogs of target analytes) should be incorporated to correct for injection volume variations and matrix effects.
The choice of drift gas requires careful consideration, with nitrogen generally providing higher resolving power while dried air may offer practical advantages for field deployment. Gas purification systems are essential for removing moisture and hydrocarbon contaminants that can compromise ionization efficiency and create elevated background signals. For portable applications, compact, integrated gas generators represent a practical alternative to compressed gas cylinders.
Sample introduction materials must be selected based on compatibility with target analytes, with inert materials such as deactivated silica or specific metal alloys preferred for reactive compounds. The use of appropriate seals, septa, and connectors minimizes the introduction of contaminants that can interfere with analysis, particularly for trace-level applications.
Comparative Performance in On-site vs. Laboratory Applications
The optimization priorities for GC-IMS parameters differ significantly between laboratory-based systems and portable instruments designed for on-site screening. Laboratory systems typically prioritize maximum resolution and sensitivity, often at the expense of analysis time and operational simplicity. In contrast, portable systems must balance analytical performance with practical constraints including power consumption, gas supply limitations, ruggedness, and analysis speed.
For on-site applications, parameter optimization often emphasizes rapid analysis cycles, with higher carrier gas flows and faster temperature ramps in the GC dimension. The IMS drift tube temperature may be maintained at a moderate setting to balance power consumption against analytical performance for the specific target compounds. Injection volumes may be increased to compensate for the reduced sensitivity resulting from these compromised conditions, though careful attention must be paid to maintaining linear response and avoiding detector saturation.
Laboratory-based GC-IMS systems can implement more comprehensive parameter optimization, with longer analysis times allowing for improved GC separation through slower temperature programming and optimized carrier gas flows. The availability of stable power and gas supplies permits operation at higher IMS temperatures when analyzing high-boiling compounds, providing superior peak shapes and mobility resolution. The controlled laboratory environment also facilitates the use of smaller injection volumes while maintaining adequate sensitivity, reducing the likelihood of non-linear detector response.
Despite these differences, both application scenarios benefit from the fundamental advantages of GC-IMS technology, including high sensitivity for volatile compounds, minimal sample preparation requirements, and the orthogonal separation provided by the two-dimensional analytical approach. The optimal parameter set for any application must ultimately reflect the specific analytical requirements, including the complexity of the sample matrix, the concentration range of target analytes, and the required throughput.
The optimization of carrier/drift gas flows, temperature regimes, and injection volume represents a critical aspect of GC-IMS method development that directly influences analytical performance across diverse application scenarios. The systematic approach to parameter optimization outlined in this guide provides a framework for balancing the often-competing demands of sensitivity, resolution, analysis speed, and operational practicality. Recent technological advancements, particularly the development of high-temperature IMS systems capable of operating at up to 180°C, have significantly expanded the application range of GC-IMS to include higher-boiling compounds that previously presented analytical challenges [51] [52].
For researchers focused on rapid on-site screening applications, the parameter optimization strategy must carefully balance analytical performance with practical constraints related to portability, power consumption, and operational simplicity. The comparative data presented in this guide provides a foundation for making informed decisions regarding these trade-offs. As GC-IMS technology continues to evolve, with ongoing improvements in instrumental design and data processing capabilities, the application space for this powerful analytical technique will undoubtedly expand, further solidifying its position as a valuable tool for both laboratory and field-based analysis of volatile compounds.
Gas Chromatography-Ion Mobility Spectrometry (GC-IMS) has emerged as a powerful analytical technique for rapid, sensitive detection of volatile organic compounds (VOCs), finding applications in food authenticity, clinical diagnostics, and forensic analysis. Despite its advantages in speed, sensitivity, and portability, GC-IMS faces a significant identification hurdle: the lack of comprehensive, universally accepted reference databases. Unlike Gas Chromatography-Mass Spectrometry (GC-MS), which benefits from extensive spectral libraries, GC-IMS compound identification typically requires comparison with reference standards, making the analysis of complex samples time-consuming and challenging. This comparison guide evaluates current strategies to overcome this limitation, providing researchers with practical approaches to enhance compound identification confidence in GC-IMS analyses.
Head-to-Head: GC-IMS Versus GC-MS Database Availability
Table 1: Comparison of GC-IMS and GC-MS Database Characteristics
| Feature | GC-IMS | GC-MS |
|---|---|---|
| Database Availability | No universally available reference database; identification must be performed individually for each study [18] | Extensive global mass spectral databases (e.g., NIST) for compound identification [18] |
| Identification Approach | Comparison of retention and drift times against reference standards [54] | Library matching of electron ionization (EI) mass spectra [54] |
| Sensitivity | Approximately ten times more sensitive than MS, with limits of detection in the picogram/tube range [18] | High sensitivity but generally lower than IMS; maintains linearity over three orders of magnitude [18] |
| Portability | High; operates at atmospheric pressure, enabling field applications [43] | Limited; requires vacuum systems, primarily laboratory-based [43] |
| Cost & Maintenance | Lower cost; simpler design with minimal maintenance [43] | Higher cost; complex instrumentation with regular maintenance needs [43] |
The core challenge stems from fundamental differences in detection principles. While MS separates ions by their mass-to-charge ratio in a vacuum, IMS separates ions based on their mobility in a buffer gas at atmospheric pressure, producing drift time spectra that are highly dependent on instrument parameters and experimental conditions [43]. This instrumentation difference has hindered the development of standardized, transferable databases for GC-IMS.
Strategic Approaches to Overcoming Database Limitations
Direct Hyphenation with Mass Spectrometry
The most effective approach to overcoming database limitations involves directly coupling GC-IMS with mass spectrometry, creating a TD-GC-MS-IMS system that leverages the strengths of both detectors [18]. This configuration uses a splitter to direct separated analytes to both detectors simultaneously, enabling:
- Reliable identification of unknowns detected by IMS through mass spectral databases [18]
- Nearly identical retention times in both detectors, ensuring accurate correlation [18]
- Enhanced analytical information from complementary detection principles [54]
Table 2: Performance Metrics of TD-GC-MS-IMS Systems
| Parameter | IMS Performance | MS Performance |
|---|---|---|
| Long-term Stability (16 months) | Signal intensity RSD: 3-13%; Drift time deviations: 0.49-0.51% [18] | Not explicitly stated in studies |
| Sensitivity | ~10x more sensitive than MS; LOD in picogram/tube range [18] | Lower sensitivity but broader linear range [18] |
| Linearity Range | 1 order of magnitude (extends to 2 orders with linearization) [18] | 3 orders of magnitude (up to 1000 ng/tube) [18] |
| Reproducibility | Precision ranges of 2.2-5.3% for sampling method [18] | Precision ranges of 3.0-7.6% for sampling method [18] |
Experimental evidence demonstrates that this hyphenated approach successfully identifies breath biomarkers such as ethanol, isoprene, and acetone, while also providing a standardized framework for clinical, environmental, and industrial VOC analysis [18].
Advanced Data Processing and Chemometric Workflows
When direct MS hyphenation isn't feasible, sophisticated data processing strategies can extract meaningful information from GC-IMS data:
Dimensionality Reduction Techniques: Principal Component Analysis (PCA) and Partial Least Squares (PLS) are effectively employed to process the high-dimensional data generated by GC-IMS, which can contain millions of data points [55]. PLS particularly outperforms PCA as a preprocessing step for supervised learning because it actively considers class/regression labels [55].
Non-Targeted Screening (NTS): Instead of identifying individual compounds, NTS approaches use complete spectral fingerprints for sample classification and discrimination, bypassing the need for comprehensive compound identification [43]. This strategy has proven effective for food authenticity control, quality monitoring, and clinical diagnostics [1] [43].
Specialized Software Tools: The GCIMS R package provides a comprehensive workflow for processing raw GC-IMS data, including denoising, baseline correction, spectral and chromatographic alignment, peak detection, and peak clustering to produce a peak table ready for multivariate analysis [47].
Standardized Sampling and Linearization Methods
Standardization of sampling protocols enhances reproducibility and comparability across studies:
- Thermal Desorption Tubes: Using a mobile flow- and temperature-controlled sampling unit for TD tubes enables standardized application for both gaseous and liquid samples, improving reproducibility [18].
- Linearization Strategies: Implementing linearization approaches extends the calibration range of IMS from one to two orders of magnitude, improving quantification capabilities [18].
Diagram Title: Strategic Framework to Overcome GC-IMS Database Limitations
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for GC-IMS Analysis
| Item | Function | Application Notes |
|---|---|---|
| Thermal Desorption Tubes | Capture and concentrate VOCs from air or other matrices [18] | Multiple adsorbent materials for different volatility ranges; enable off-site sampling [18] |
| Reference Standards | Compound identification via retention and drift time comparison [18] | Purity ≥95%; prepare stock solutions in methanol or appropriate solvents [18] |
| Tritium Ionization Source | Ionizes VOCs through proton-transfer reactions [43] | Low activity (300 MBq) exempt from authorization in EU; forms reactant ion peaks (RIP) [43] |
| Chemical Standards | System calibration and performance verification [18] | Ketones, aldehydes, alcohols; used for long-term stability assessment [18] |
| Drift Gas | Inert buffer gas for ion separation in drift tube [43] | Typically nitrogen; requires precise moisture and temperature control [43] |
Experimental Protocols for Method Validation
Protocol 1: TD-GC-MS-IMS System Performance Assessment
Based on established methodology [18], this protocol validates system performance:
- Standard Preparation: Prepare three stock solutions (alcohols, aldehydes, ketones) in methanol with reference substances of ≥95% purity.
- System Configuration: Implement a splitter after GC separation to direct effluent to both IMS and MS detectors simultaneously.
- Long-term Stability Test: Conduct measurements over extended periods (e.g., 16 months) with regular injections of ketone standards.
- Performance Metrics: Calculate relative standard deviations for signal intensities (target: 3-13%), retention time deviations (target: 0.10-0.22%), and drift time deviations (target: 0.49-0.51%).
- Sensitivity Comparison: Determine limits of detection for both detectors using serial dilutions, expecting IMS to be approximately ten times more sensitive than MS.
Protocol 2: Non-Targeted Screening with Chemometric Analysis
For samples where comprehensive identification isn't required [55] [43]:
- Sample Analysis: Collect GC-IMS fingerprints from all sample groups under standardized conditions.
- Data Preprocessing: Use the GCIMS R package for denoising, baseline correction, alignment, and peak detection [47].
- Dimensionality Reduction: Apply PLS with Variable Importance in Projection (VIP) scores to identify features most relevant to sample classification [55].
- Model Validation: Implement cross-validation and test with independent sample sets to ensure model robustness.
- Marker Investigation: Correlate significant features with sample properties, potentially identifying candidate markers for future targeted analysis.
While GC-IMS faces significant database limitations compared to GC-MS, the strategic approaches outlined provide effective pathways to overcome this hurdle. Direct hyphenation with mass spectrometry offers the most comprehensive solution by leveraging existing MS databases, while advanced data processing and standardized protocols enable robust analysis when hyphenation isn't feasible. The choice between these strategies depends on specific application requirements, available resources, and analytical goals. As GC-IMS technology continues to evolve, these approaches provide researchers with practical methodologies to harness the technique's superior sensitivity and portability while maintaining confidence in compound identification.
Benchmarking Performance: Rigorous Validation of GC-IMS Against GC-MS
In the evolving landscape of analytical chemistry, the choice between Gas Chromatography-Ion Mobility Spectrometry (GC-IMS) and Gas Chromatography-Mass Spectrometry (GC-MS) represents a critical decision point for researchers. GC-IMS has emerged as a powerful technique for rapid on-site screening, offering exceptional sensitivity and speed. In contrast, GC-MS remains the gold standard for laboratory-based confirmation, providing robust quantification and identification. This comparison guide objectively examines the performance characteristics of both techniques, focusing on their limits of detection (LOD) and linear dynamic ranges to inform method selection for various analytical scenarios.
How It Works: GC-IMS vs. GC-MS
GC-IMS Operational Principles
GC-IMS combines two separation techniques to analyze volatile organic compounds (VOCs). First, gas chromatography separates compounds based on their partitioning between a mobile gas phase and a stationary phase column [14]. The separated components then enter the ion mobility spectrometer, where they are ionized, typically using a radioactive source such as tritium or ⁶³Ni [6]. These ions are subjected to a weak electric field in a drift tube filled with an inert buffer gas, separating them based on their size, shape, and charge as they drift toward a detector [14] [56]. The result is a two-dimensional plot of GC retention time versus IMS drift time, creating a unique "fingerprint" for each compound [4].
GC-MS Operational Principles
GC-MS similarly begins with gas chromatography to separate volatile compounds. However, the detection mechanism differs significantly. Eluting compounds enter the mass spectrometer, where they are ionized by electron impact or chemical ionization under high vacuum conditions [6]. The resulting ions are then separated in the mass analyzer based on their mass-to-charge ratio (m/z) [56]. This process generates a mass spectrum that serves as a molecular signature, which can be compared against extensive reference libraries for confident identification [6].
Quantitative Performance Comparison
Limits of Detection (LOD)
The exceptional sensitivity of GC-IMS represents one of its most significant advantages for trace analysis. A comprehensive 2025 study directly compared the quantification performance of thermal desorption (TD) GC-IMS and GC-MS, revealing that IMS was approximately ten times more sensitive than MS, achieving limits of detection in the picogram per tube range [57] [6]. This exceptional sensitivity makes GC-IMS particularly valuable for applications where detecting trace-level compounds is critical, such as in security screening for explosives or medical diagnostics using breath biomarkers [58] [1].
Linear Dynamic Range
While GC-IMS offers superior sensitivity, GC-MS provides a significant advantage in linear dynamic range. The same 2025 study demonstrated that MS exhibited a broader linear range, maintaining linearity over three orders of magnitude (up to 1000 ng/tube) [57] [6]. In contrast, IMS retained linearity for only one order of magnitude (e.g., 0.1 to 1 ng/tube for pentanal) before transitioning into a logarithmic response [57] [6]. This limitation can be partially mitigated through linearization strategies, which have been shown to extend the IMS calibration range from one to two orders of magnitude [6].
Table 1: Direct Comparison of Key Quantitative Performance Metrics
| Parameter | GC-IMS | GC-MS |
|---|---|---|
| Typical Limit of Detection | Picogram/tube range [57] [6] | Approximately 10x higher than IMS [57] [6] |
| Linear Dynamic Range | 1-2 orders of magnitude (with linearization) [6] | 3 orders of magnitude (up to 1000 ng/tube) [57] [6] |
| Long-term Signal Intensity RSD | 3% to 13% over 16 months [57] [6] | Not explicitly stated in studies |
| Retention Time Stability | 0.10% to 0.22% deviation [57] [6] | Typically <2% deviation [6] |
| Drift Time Stability | 0.49% to 0.51% deviation [57] [6] | Not applicable |
Table 2: Technique Selection Guide Based on Analytical Needs
| Analytical Requirement | Recommended Technique | Rationale |
|---|---|---|
| Trace-level detection | GC-IMS | Superior sensitivity with picogram-level LOD [57] [6] |
| Wide concentration range quantification | GC-MS | Broader linear dynamic range (3 orders of magnitude) [57] [6] |
| Rapid on-site screening | GC-IMS | Portable configurations available; faster analysis times [14] [1] |
| Unknown compound identification | GC-MS | Extensive mass spectral libraries available [6] |
| Isomer differentiation | GC-IMS | Better separation of structural isomers based on shape [56] |
| Long-term reproducibility | Both | Both demonstrate good stability, with IMS showing 3-13% RSD over 16 months [6] |
Experimental Protocols and Methodologies
Standardized TD-GC-MS-IMS Framework
Recent research has developed integrated approaches to leverage the strengths of both techniques. A 2025 study established a standardized thermal desorption GC-MS-IMS framework using a mobile flow- and temperature-controlled sampling unit for TD tubes [6]. This system was designed to introduce both gaseous and liquid samples, with analytes directed to both detectors after GC separation via a simple splitter, ensuring nearly identical retention times [6].
Key Experimental Parameters
- Long-term Stability Assessment: Conducted over 16 months with 156 measurement days using ketones as reference compounds [6]
- Calibration Approach: Employed liquid external standards prepared in methanol, with stock solutions for alcohols, aldehydes, and ketones [6]
- Linearization Strategy: Implemented mathematical correction to extend the IMS calibration range from one to two orders of magnitude [6]
- System Suitability: Verified using relative standard deviations for signal intensities (3-13%), retention time (0.10-0.22%), and drift time (0.49-0.51%) [6]
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Materials for TD-GC-IMS Experiments
| Item | Function | Application Notes |
|---|---|---|
| Thermal Desorption Tubes | Sample collection and concentration | Filled with adsorbent material; trap VOCs from air or other matrices [6] |
| Sorbent Materials | VOC retention and release | Choice depends on target compounds; determines boiling point range (150°C to 250°C) [6] |
| Methanol (GC Ultra Grade) | Solvent for standard preparation | Purity ≥99.9%; used for preparing calibration solutions [6] |
| Ketone Standards | System performance verification | Used for long-term stability assessment (e.g., 2-butanone) [6] |
| Aldehyde Standards | Calibration and quantification | Examples: propanal, butanal, pentanal, hexanal [6] |
| Alcohol Standards | Calibration and quantification | Examples: 1-propanol, 1-butanol, 1-pentanol, 1-hexanol [6] |
| Drift Gas | IMS buffer gas | Clean air or nitrogen; requires moisture trap to control humidity effects [4] [6] |
| Dopants | Selective ionization | Enhance detection of specific compound classes (e.g., C₂Cl₆ for explosives) [58] |
Operational Characteristics and Practical Considerations
Analysis Time and Portability
GC-IMS offers significant advantages in analysis speed, with typical completion times of 3 to 5 minutes [14]. The technique is also more amenable to miniaturization, with portable systems available for field deployment [14] [59]. This makes GC-IMS particularly valuable for applications requiring rapid on-site decisions, such as security screening, environmental monitoring, and clinical diagnostics [1].
Matrix Effects and Selectivity
A significant consideration for GC-IMS is its susceptibility to matrix effects, where the ionization of target analytes can be suppressed by co-eluting compounds [4]. This challenge is mitigated by the preceding GC separation, which reduces component competition for ionization [4]. GC-MS generally offers better selectivity in complex matrices, particularly when using tandem mass spectrometry approaches.
Identification Capabilities
GC-MS maintains a substantial advantage in compound identification due to the availability of extensive mass spectral libraries [6]. In contrast, IMS lacks universally available reference databases, making compound identification more challenging [6]. This limitation can be addressed by coupling IMS with MS in parallel systems, allowing unknown compounds in IMS data to be identified using mass spectrometric libraries [6].
The choice between GC-IMS and GC-MS ultimately depends on specific analytical requirements. GC-IMS excels in scenarios demanding maximum sensitivity, rapid analysis, and portability for on-site screening. Its picogram-level detection limits make it invaluable for trace-level analysis when the concentration range is limited. Conversely, GC-MS remains superior for applications requiring broad linear dynamic range, confident compound identification across wide concentration ranges, and laboratory-based confirmation. The emerging trend of combined GC-MS-IMS systems represents a promising direction, leveraging the complementary strengths of both techniques to provide comprehensive analytical solutions with both exceptional sensitivity and robust quantification capabilities.
Gas Chromatography-Ion Mobility Spectrometry (GC-IMS) represents a significant technological advancement for the analysis of volatile organic compounds (VOCs), positioning itself as a robust alternative to traditional laboratory-based methods like GC-Mass Spectrometry (GC-MS). This comparison guide objectively assesses the quantitative performance of GC-IMS against established alternatives, with particular focus on its applicability for rapid on-site screening. The demand for analytical techniques that offer both high sensitivity and portability has increased substantially in fields ranging from clinical diagnostics to environmental monitoring and food safety. GC-IMS addresses this need by combining the superior separation capabilities of gas chromatography with the high sensitivity and rapid detection of ion mobility spectrometry, all while operating at ambient pressure and offering significantly reduced operational complexity compared to GC-MS [16].
The core thesis framing this assessment posits that GC-IMS delivers quantitative performance comparable to laboratory-bound methods for specific applications, while offering distinct advantages in speed, cost-efficiency, and operational flexibility that make it particularly suitable for decentralized analysis. Understanding its quantitative rigor—specifically long-term stability, reproducibility, and linearity—is essential for researchers and drug development professionals evaluating appropriate analytical platforms for their specific volatilomic applications. This guide provides experimental data and comparative metrics to inform these critical technology selection decisions.
Experimental Comparison: GC-IMS Versus GC-MS Performance Metrics
Core Performance Metrics
A comprehensive 16-month study evaluating a thermal desorption GC-MS-IMS system provides critical quantitative data for direct comparison between these technologies. The assessment, conducted over 156 measurement days using ketones as test analytes, revealed distinct performance characteristics for each detection method [18] [57].
Table 1: Comparative Performance Metrics of GC-IMS and GC-MS
| Performance Parameter | GC-IMS | GC-MS |
|---|---|---|
| Long-Term Signal Intensity RSD | 3% to 13% | Similar precision range reported |
| Retention Time Deviations | 0.10% to 0.22% | Nearly identical (shared GC separation) |
| Drift Time Deviations | 0.49% to 0.51% | Not applicable |
| Approximate Sensitivity | ~10x higher than MS | Baseline for comparison |
| Typical Limits of Detection | Picogram/tube range | Higher than IMS (exact multiple not specified) |
| Linear Range | 1 order of magnitude (e.g., 0.1-1 ng/tube for pentanal) | 3 orders of magnitude (up to 1000 ng/tube) |
| Response Beyond Linear Range | Logarithmic | Maintained linearity |
| Effective Linear Range with Linearization | 2 orders of magnitude | Not required |
The data reveals a fundamental trade-off: IMS provides approximately ten times higher sensitivity than MS, achieving detection limits in the picogram per tube range. However, MS exhibits a substantially broader linear range, maintaining linearity over three orders of magnitude compared to just one order of magnitude for IMS [18]. This distinction is critical for method selection—IMS is superior for trace-level detection, while MS is more suitable for applications requiring quantification across wide concentration ranges.
Long-Term Stability Assessment
The long-term stability of GC-IMS is particularly notable for on-site applications where instrument recalibration may be challenging. The reported relative standard deviations for signal intensities (3-13%), retention times (0.10-0.22%), and drift times (0.49-0.51%) over the 16-month period demonstrate remarkable system stability [18]. This level of reproducibility is comparable to laboratory-based systems and confirms the viability of GC-IMS for extended field deployment and monitoring applications.
Diagram 1: TD-GC-MS-IMS experimental workflow for comparative performance assessment.
Detailed Experimental Protocols for Performance Validation
TD-GC-MS-IMS System Configuration and Operation
The fundamental experimental setup for comparative assessment utilizes a thermal desorption gas chromatography system coupled to both mass spectrometry and ion mobility spectrometry detectors (TD-GC-MS-IMS). This configuration enables simultaneous detection and direct comparison of both techniques from the same analytical separation [18]. The system employs a simple splitter to direct analytes after GC separation to both detectors, ensuring nearly identical retention times and eliminating separation variability from the comparison [18].
Critical to the quantitative reliability of this method is the standardized sampling system utilizing thermal desorption tubes. These tubes capture and concentrate VOCs from various matrices, which are then released through controlled heating and introduced into the chromatographic system. A mobile flow- and temperature-controlled sampling unit was developed specifically for this application to ensure reproducible analyte collection for both gaseous and liquid samples [18]. This standardized sampling approach is essential for achieving the reported precision metrics.
For quantitative calibration, reference substances with purity ≥95% are typically used. Researchers often prepare stock solutions grouped by chemical class (alcohols, aldehydes, ketones), with methanol (99.9% GC Ultra Grade) serving as the solvent [18]. While international standards permit both liquid and vapor phase standards for TD calibration, liquid standard introduction is more cost-effective, though it requires strict control of temperature and gas flow to ensure reproducible adsorption onto the sorbent material [18].
Linearization Strategy for Extended IMS Quantification
A significant methodological advancement for GC-IMS quantification involves a linearization strategy to extend its limited calibration range. While IMS detection naturally transitions to a logarithmic response beyond approximately one order of magnitude, researchers have implemented mathematical approaches to extend the effective linear range to two orders of magnitude [18]. This expansion substantially improves the practical utility of GC-IMS for quantitative analysis without sacrificing its inherent sensitivity advantages.
The experimental protocol for this approach involves:
- Establishing a traditional calibration curve across the limited linear range (e.g., 0.1-1 ng/tube for pentanal)
- Characterizing the logarithmic response pattern at higher concentrations
- Applying algorithm-based correction to linearize the response
- Validating the linearized response against known standards
This linearization process enables GC-IMS to maintain quantitative accuracy across a more practically useful concentration range while preserving its superior sensitivity characteristics compared to GC-MS.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Research Reagents and Materials for TD-GC-MS-IMS Experiments
| Reagent/Material | Specifications | Function in Experimental Protocol |
|---|---|---|
| Thermal Desorption Tubes | Multiple adsorbent beds for VOC retention | Sample collection, concentration, and introduction into the analytical system |
| Reference Standards | Purity ≥95% (aldehydes, ketones, alcohols) | Method calibration, quantification, and performance validation |
| Methanol Solvent | 99.9% GC Ultra Grade | Preparation of calibration solutions and stock standards |
| Ketone Calibration Mixtures | Certified reference materials | Long-term stability assessment (e.g., 16-month precision study) |
| Quality Control (QC) Samples | Pooled from all test samples | Monitoring instrument performance and signal correction |
| Molecular Sieves | 13X form, 8-12 mesh | Purification of carrier/drift gases and humidity control |
Green Analytical Chemistry and Practical Implementation
Environmental and Operational Advantages
Beyond pure performance metrics, GC-IMS offers significant practical advantages that align with the principles of Green Analytical Chemistry (GAC). Compared to GC-MS, which requires high vacuum conditions, substantial energy inputs, and often uses helium—a non-renewable resource—as carrier gas, GC-IMS operates at ambient pressure and can utilize air as carrier gas [16]. This translates to reduced operational costs and environmental impact, particularly important for extended on-site deployments.
Modern GC-IMS systems feature smaller footprints than comparable GC-MS instrumentation, enhancing their suitability for space-constrained environments [16]. While IMS systems commonly employ radioactive ionization sources (typically tritium, ≤100 MBq), these are sealed, low-dose sources that can be recycled for reuse by the manufacturer, minimizing environmental concerns [16].
Addressing Technical Limitations
The primary limitation of GC-IMS—its narrower linear dynamic range compared to GC-MS—can be mitigated through the linearization strategies previously discussed. Additionally, the technique's historical lack of universal reference databases (unlike the extensive libraries available for GC-MS) can be addressed by parallel MS-IMS detection, which allows unknown compounds detected by IMS to be reliably identified using mass spectral databases [18].
Diagram 2: Analytical technique selection guide based on application requirements.
The quantitative assessment of GC-IMS reveals a technique with distinct advantages for specific application scenarios. Its superior sensitivity, demonstrated by detection limits in the picogram range and approximately 10-fold greater sensitivity than GC-MS, combined with excellent long-term stability (3-13% RSD over 16 months), positions it as an ideal platform for rapid on-site screening applications where trace-level detection is paramount [18] [57].
While GC-MS maintains advantages in linear dynamic range and compound identification through extensive libraries, the development of linearization strategies for IMS quantification and the ability to couple both detectors in parallel systems effectively address these limitations. For researchers and drug development professionals requiring decentralized analysis capabilities without sacrificing quantitative rigor, GC-IMS represents a compelling alternative to traditional laboratory-based methods.
The experimental data and comparative metrics presented in this guide provide a foundation for informed technology selection based on specific application requirements, performance expectations, and operational constraints. As GC-IMS technology continues to evolve and its methodological approaches mature, its adoption across diverse volatilomic applications is likely to accelerate, particularly in fields where the balance between analytical performance and practical utility is paramount.
In the evolving landscape of analytical chemistry, a fundamental tension exists between the need for rapid results and the requirement for unambiguous compound identification. This challenge is particularly acute in fields such as forensic science, food safety, and clinical diagnostics where timely decisions must be balanced against analytical confidence. Gas Chromatography-Ion Mobility Spectrometry (GC-IMS) has emerged as a powerful technology promising to bridge this gap, offering rapid on-site analysis capabilities while maintaining reasonable specificity.
This guide provides a direct comparison between GC-IMS and established laboratory techniques, focusing specifically on analysis speed and compound identification confidence. We examine experimental data across multiple application domains to provide researchers, scientists, and drug development professionals with objective performance metrics for selecting appropriate analytical methodologies based on their specific needs for timeliness versus certainty.
Analytical Techniques Compared
GC-IMS (Gas Chromatography-Ion Mobility Spectrometry): This technique combines the separation power of gas chromatography with the detection capability of ion mobility spectrometry. After GC separation, molecules are ionized and their drift time through an electric field in a carrier gas is measured, creating two-dimensional separation (retention time and drift time) for compound identification [38] [12]. Modern hyper-fast GC-IMS systems can complete analyses in 20-30 seconds while maintaining low ppbv detection limits [60].
GC-MS (Gas Chromatography-Mass Spectrometry): The established laboratory standard that separates compounds via GC followed by mass-based identification using mass spectrometry. This technique provides high specificity through mass spectral matching against reference libraries but typically requires longer analysis times and laboratory infrastructure [61].
GC×GC-MS (Comprehensive Two-Dimensional Gas Chromatography-Mass Spectrometry): An advanced separation technique that employs two different GC columns in sequence, significantly enhancing peak capacity and separation power over conventional GC-MS. This results in superior specificity but with increased analytical complexity and time requirements [62] [61].
Key Performance Metrics
The comparison between these techniques primarily revolves around several critical performance parameters:
- Analysis Time: Total duration from sample introduction to result availability
- Detection Limits: Lowest concentration of analyte that can be reliably detected
- Identification Confidence: Certainty of compound identification, often measured by false positive/negative rates
- Peak Capacity: Number of distinct compounds that can be separated in a single analysis
- Portability: Suitability for field deployment versus laboratory requirement
Direct Performance Comparison
Quantitative Performance Metrics
Table 1: Direct Comparison of Analytical Technique Performance Characteristics
| Performance Parameter | GC-IMS | GC-MS | GC×GC-MS |
|---|---|---|---|
| Typical Analysis Time | 30 seconds - 5 minutes [60] [38] | 15-60 minutes [61] | 30-90 minutes [61] |
| Detection Limits | Low ppbv range (single-digit) [12] [60] | Low ppb-ppt range [61] | Low ppb-ppt range [61] |
| Identification Basis | Retention time + drift time [12] | Retention time + mass spectrum [61] | 2D retention time + mass spectrum [61] |
| Peak Capacity | Moderate (two-dimensional separation) [38] | High [61] | Very high (3-5× GC-MS) [61] |
| Portability | High (portable systems available) | Low (laboratory-based) | Low (laboratory-based) |
| Typical Applications | On-site screening, rapid quality control [38] [12] | Definitive confirmation, reference methods [63] [61] | Complex mixtures, untargeted analysis [62] [61] |
Application-Specific Performance
Table 2: Performance in Specific Application Domains
| Application Domain | Analytical Technique | Key Findings | Reference |
|---|---|---|---|
| Mold Detection in Tobacco | GC-IMS | Identified 4 early-warning biomarkers; 1.3-1.5-fold increases in early mold stages; Analysis complete in minutes | [38] |
| Clandestine Drug Laboratory Detection | GC-IMS | Detected precursors at single-digit ppbv levels; Reliable identification via retention time + ion mobility | [12] |
| Human Serum Metabolomics | GC-MS vs. GC×GC-MS | GC×GC-MS detected 3× more peaks vs. GC-MS at SNR ≥ 50; 23 significant biomarkers (GC-MS) vs. 34 (GC×GC-MS) | [61] |
| Synthetic Cannabinoid Screening | Portable Electrochemical vs. GC-MS | 83% accuracy for on-site device vs. GC-MS confirmation; Results in <1 minute vs. hours/days | [63] |
| Hop Variety Analysis | Hyper-fast GC-IMS | Complete analysis in 20 seconds; Resolved peaks with FWHM of 140 ms | [60] |
Experimental Protocols and Methodologies
GC-IMS Analysis of Mold Biomarkers
The identification of early-warning biomarkers for mold contamination in cigar tobacco leaves exemplifies standardized GC-IMS protocols [38]:
- Sample Preparation: Tobacco samples (1.0 g) were placed in headspace sampling vials, sealed, and incubated at 80°C for 15 min with agitation at 500 rpm
- GC Conditions: MXT-WAX column (15 m × 0.53 mm, 1.0 µm) at 60°C; Nitrogen carrier gas with programmed flow: 0-2 min at 2.0 mL/min, 2-8 min increased to 10.0 mL/min, 8-10 min increased to 100.0 mL/min, held for 10 min
- IMS Conditions: Ionization source not specified in detail; Detection via drift time measurement
- Data Analysis: 72 VOCs detected; Four compounds (2-methyl-1-butanol-M, 2-methyl-1-butanol-D, 2-propanone, and 1-penten-3-ol) identified as early-warning biomarkers through VIP > 1 and P < 0.05
- Analysis Time: Complete analysis achieved within approximately 20 minutes including sample preparation
Comparative Metabolomics Protocol
A detailed comparison between GC-MS and GC×GC-MS for serum metabolomics highlights methodological considerations for laboratory-based techniques [61]:
- Sample Preparation: 100 µL serum added to 1 mL ice-cold methanol/chloroform (3:1) with internal standards; Vortexed and centrifuged; Supernatant dried under N₂; Derivatized with methoxyamine in pyridine (90 min at 30°C) followed by MSTFA with 1% TMCS (60 min at 70°C)
- GC-MS Analysis: 60 m × 0.25 mm DB-5 ms UI column; 1.0 mL/min helium; Temperature program: 60°C for 1 min, then 5°C/min to 300°C, holding 12 min; Mass range: m/z 45-1000
- GC×GC-MS Analysis: Primary column identical to GC-MS; Secondary column: 1 m × 0.25 mm DB-17 ms; Modulator period: 2.5 s; Secondary oven +10°C relative to primary; Mass acquisition rate: 200 spectra/s
- Quality Control: Pooled QC samples analyzed after every 9 biological samples; Randomized analysis order
- Data Processing: LECO ChromaTOF for peak detection; MetPP for peak merging and alignment; NIST11 and Fiehn libraries for metabolite identification
Hyper-Fast GC-IMS Protocol
Advanced GC-IMS systems have dramatically reduced analysis times while maintaining performance [60]:
- Instrument Design: Dual drift tube IMS in axial configuration with directed sample gas flow; Effective detector volume: 40 µL; Isothermal heating at 120°C
- Drift Tube Specifications: Length 41 mm; Drift voltage 2.7 kV; Repetition rate: 100 Hz; Resolving power: Rp = 60
- Chromatographic Performance: Capable of resolving GC peaks with full width at half maximum of only 140 ms
- Analysis Time: Complete analyses (including hops varieties and explosives mixture) achieved within 20-30 seconds total run time including cooling
Workflow and Decision Pathways
Figure 1: Analytical Technique Selection Pathway Based on Speed and Specificity Requirements
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagents and Materials for GC-IMS and Comparative Techniques
| Item | Function/Purpose | Example Specifications | Application |
|---|---|---|---|
| MXT-WAX GC Column | Primary separation column for polar compounds | 15 m × 0.53 mm, 1.0 µm film [38] | GC-IMS analysis |
| DB-5 ms UI GC Column | Standard non-polar/phenyl mid-polar column | 60 m × 0.25 mm, 0.25 µm film [61] | GC-MS and GC×GC-MS |
| DB-17 ms GC Column | Secondary column for 2D separation | 1 m × 0.25 mm, 0.25 µm film [61] | GC×GC-MS (2nd dimension) |
| Methoxyamine in Pyridine | Derivatization reagent for carbonyl groups | 20 mg/mL concentration [61] | Sample preparation for GC |
| MSTFA with 1% TMCS | Silylation reagent for hydroxyl and amine groups | N-methyl-N-(trimethylsilyl) trifluoroacetamide [61] | Sample preparation for GC |
| Alkane Retention Index Standard | Retention time calibration | C10-C40 alkanes [61] | Method standardization |
| Boron-Doped Diamond Electrode | Electrochemical sensor for portable devices | Commercial BDDE [63] | Portable screening |
| Quality Control Reference Materials | Method validation and quality control | Pooled serum samples [61] | Analytical QC |
Discussion and Technical Considerations
Interpretation of Comparative Data
The experimental data reveals a clear trade-off between analysis speed and compound identification confidence. GC-IMS provides dramatically faster analysis (30 seconds to 5 minutes) compared to laboratory techniques (15-90 minutes), while maintaining sufficient specificity for many screening applications [38] [60]. However, this speed advantage comes at the cost of reduced peak capacity and identification certainty compared to MS-based techniques.
The comprehensive comparison of GC-MS and GC×GC-MS for serum metabolomics demonstrates that while GC×GC-MS provides 3× greater peak detection capability and identifies more significant biomarkers (34 vs. 23), it requires more complex instrumentation, longer analysis times, and advanced data processing capabilities [61]. This makes it unsuitable for rapid screening but superior for discovery-phase research.
Confidence in Compound Identification
A critical consideration in technique selection is the fundamental basis for compound identification and the associated confidence levels:
- GC-IMS identification relies on two-dimensional separation (retention time + drift time) without molecular fragmentation data, potentially leading to higher false-positive rates for complex matrices [12]
- GC-MS identification incorporates mass spectral matching, providing significantly higher confidence through fragmentation patterns and extensive reference libraries [61]
- GC×GC-MS identification combines superior chromatographic separation with mass spectral data, providing the highest confidence for complex samples but requiring the greatest analytical resources [61]
Recent advances in computational prediction of molecular properties have begun to influence identification confidence across all techniques. The concept of "reference-free" identification using predicted properties (retention time, collision cross section, mass spectra) is expanding the identifiable chemical space beyond the limitations of authentic standards [64].
Application-Based Recommendations
Based on the comparative data, technique recommendations vary significantly by application need:
- Routine Quality Control & Screening: GC-IMS provides optimal performance for high-throughput environments where rapid results outweigh maximum specificity needs [38]
- Forensic Confirmation & Regulatory Testing: GC-MS remains the preferred choice when results must withstand legal challenges or regulatory scrutiny [63]
- Research & Biomarker Discovery: GC×GC-MS offers superior performance for untargeted analysis of complex biological matrices, despite longer analysis times [61]
The choice between analytical techniques fundamentally involves balancing the competing priorities of speed and specificity. GC-IMS technologies provide remarkable analysis speed measured in seconds to minutes, making them ideal for rapid on-site screening applications where immediate results drive decision-making. In contrast, traditional GC-MS and GC×GC-MS offer significantly higher identification confidence through mass spectral verification and superior separation power, remaining essential for definitive analysis, regulatory compliance, and research applications.
This comparison demonstrates that rather than a one-size-fits-all solution, modern analytical chemistry requires a portfolio approach where technique selection is matched to specific application requirements. GC-IMS has established a firm position in the analytical toolkit for situations demanding rapid analysis, while mass spectrometry-based methods maintain their dominance when uncompromising identification confidence is required. Understanding these trade-offs enables researchers to make informed decisions that optimize both analytical efficiency and results credibility.
The analysis of volatile organic compounds (VOCs) is crucial across numerous fields, from environmental monitoring to clinical diagnostics and pharmaceutical quality control. Within this landscape, Gas Chromatography-Ion Mobility Spectrometry (GC-IMS) and Gas Chromatography-Mass Spectrometry (GC-MS) have emerged as powerful yet distinct analytical techniques. GC-IMS is renowned for its exceptional sensitivity and rapid analysis capabilities, making it ideal for rapid on-site screening. In contrast, GC-MS is a well-established laboratory-based method known for its superior compound identification and robust quantification. Rather than existing as competitors, these techniques possess a powerful, often underutilized, synergistic potential.
This guide objectively compares the performance of GC-IMS and GC-MS by examining recent experimental data. It demonstrates how their complementary strengths can be strategically implemented to create a more powerful analytical workflow. This approach leverages GC-IMS for high-speed, sensitive initial screening and uses GC-MS for definitive identification and precise quantification, thereby bridging the gap between field-based analysis and laboratory confirmation.
Technical Comparison: Operational Principles and Performance Metrics
The fundamental differences between IMS and MS detectors, coupled with their shared gas chromatography front-end, define their respective analytical niches and complementary potential.
Detector Operating Principles
- Ion Mobility Spectrometry (IMS): IMS separates ionized analyte molecules based on their size, shape, and charge as they drift through a buffer gas under the influence of an electric field. The measured drift time is characteristic of the ion. IMS typically uses chemical ionization at atmospheric pressure, making it highly sensitive but susceptible to matrix effects where co-eluting compounds can compete for ionization [4] [9].
- Mass Spectrometry (MS): MS separates ions by their mass-to-charge ratio (m/z) in a high-vacuum environment. Electron impact ionization at 70 eV is standard, providing rich, reproducible fragmentation patterns that are ideal for identifying unknown compounds by comparison with extensive standard libraries [65] [66].
Key Performance Data
The following table summarizes quantitative performance data derived from recent comparative studies.
Table 1: Performance Comparison of GC-IMS and GC-MS
| Performance Characteristic | GC-IMS | GC-MS (Benchtop) | Experimental Context |
|---|---|---|---|
| Sensitivity (Limit of Detection) | ~10x more sensitive than MS; picogram/tube range [57] | High; typically low picogram [66] | Comparative evaluation of a TD-GC-MS-IMS system [57] |
| Linear Dynamic Range | 1 order of magnitude (extendable to 2 with linearization) [57] | >3 orders of magnitude (up to 1000 ng/tube) [57] | Calibration with ketones and pentanal [57] |
| Long-Term Stability (Precision) | RSD: 3-13% (intensity), 0.10-0.22% (retention time), 0.49-0.51% (drift time) over 16 months [57] | High reproducibility; typically <10% RSD common for quantification [65] | Long-term stability assessment over 156 measurement days [57] |
| Analysis Speed | Very fast; hyper-fast GC-IMS achieves runs in <30 seconds [11] | Slower; minutes to tens of minutes per run [67] | Analysis of ketone mixes and hop varieties [11] |
| Portability | High; portable systems available, low maintenance [1] [9] | Low; primarily benchtop. Portable systems exist but with performance trade-offs [68] | Field applications for air quality and clinical diagnostics [1] [9] |
| Identification Power | Based on retention index and drift time; limited library databases [69] | Excellent; based on retention time and characteristic mass spectrum; extensive commercial libraries available [67] [66] | Identification of VOCs in food and air quality studies [69] [9] |
Experimental Protocols for Comparative Analysis
To generate the comparative data cited in this guide, researchers employ standardized methodologies. The following protocols outline a typical setup for a synergistic investigation.
Protocol 1: Long-Term Stability and Quantitative Performance Assessment
This protocol is designed to evaluate the precision, sensitivity, and linearity of an integrated GC-MS-IMS system over time [57].
- 1. System Configuration: A thermal desorption gas chromatography system coupled to both a mass spectrometer and an ion mobility spectrometer (TD-GC-MS-IMS) is used.
- 2. Sampling Unit: A mobile, flow- and temperature-controlled sampling unit for TD tubes is employed to ensure standardized application for both gaseous and liquid samples.
- 3. Stability Testing:
- Analytes: A mixture of ketones.
- Duration: Measurements are taken over 156 days across a 16-month period.
- Data Collection: Signal intensities, GC retention times, and IMS drift times are recorded for each measurement.
- Data Analysis: Relative standard deviations (RSD) are calculated for all parameters to assess long-term precision.
- 4. Sensitivity & Linearity Testing:
- Analytes: A series of VOCs, including pentanal.
- Procedure: Calibration curves are generated for both the IMS and MS detectors across a defined concentration range.
- Data Analysis: Limits of detection (LOD) are determined, and the linear range for each detector is established. A linearization strategy may be applied to the IMS data to extend its usable calibration range.
Protocol 2: On-Site Screening with Confirmatory Laboratory Analysis
This protocol leverages the strengths of both portable GC-IMS and laboratory-based GC-MS for a comprehensive field-to-lab workflow [9] [68].
- 1. On-Site Sampling:
- Equipment: Portable GC-IMS with a thermal desorption unit or SPME inlet.
- Location: Sampling is conducted at multiple points of interest (e.g., indoor environments, industrial settings).
- 2. Field Analysis:
- Procedure: Samples are analyzed directly on the portable GC-IMS.
- Output: Rapid generation of 2D fingerprint plots (drift time vs. retention time). Differential analysis of these plots quickly identifies locations with anomalous VOC signatures for further investigation.
- 3. Confirmatory Laboratory Analysis:
- Equipment: Benchtop GC-MS system.
- Sample Transfer: A subset of samples, particularly those showing anomalies in the field, are transported to the laboratory for confirmatory analysis.
- Procedure: Samples are analyzed using the GC-MS, leveraging its high resolving power and extensive spectral libraries.
- Output: Definitive identification and precise quantification of the VOCs detected in the field.
Workflow Visualization: A Synergistic Analysis Pathway
The following diagram illustrates the integrated workflow that leverages both GC-IMS and GC-MS, from sample collection to final reporting.
Essential Research Reagent Solutions
Successful implementation of the synergistic GC-IMS and GC-MS workflow relies on key reagents and materials. The following table details these essential components and their functions.
Table 2: Key Research Reagents and Materials for VOC Analysis
| Item | Function / Application | Technical Notes |
|---|---|---|
| Thermal Desorption (TD) Tubes | Sample collection and pre-concentration of VOCs from air/gas streams [57] [68]. | Typically filled with sorbents like Tenax TA; require conditioning before use [68]. |
| SPME Fibers | Needle-based microextraction for headspace sampling of liquids or solids [65] [68]. | Various coatings (e.g., PDMS-DVB) available for different analyte selectivity. |
| High-Purity Drift Gas | The neutral gas that fills the drift region of the IMS, enabling ion separation [4] [69]. | Nitrogen or clean, dry air is used; often requires a moisture trap [4] [69]. |
| Derivatization Agents | Chemically modify non-volatile or reactive analytes to create volatile, stable derivatives for GC analysis [65]. | Used in GC-MS for analyzing compounds like sulfonic acid esters (e.g., with pentafluorothiophenol) [65]. |
| Internal Standards | Added in known amounts to correct for sample loss and instrumental variability, improving quantitative accuracy [65]. | Isotopically labeled analogs of the target analytes are ideal for both GC-MS and GC-IMS quantification [65]. |
| Calibration Standards | Used to establish instrument response and create calibration curves for quantification [57] [65]. | For GC-IMS, a series of ketones (C4–C9) are commonly used to calculate retention indices [69]. |
Application Scenarios: From Screening to Confirmation
The complementary nature of GC-IMS and GC-MS makes them ideally suited for a tiered analytical approach in various fields.
- Clinical Diagnostics and Breath Analysis: GC-IMS is transformative for rapid, non-invasive breath analysis, allowing high-throughput screening for diseases by detecting VOC biomarkers in seconds to minutes [1]. Suspicious samples or those indicating a disease signature can be collected in a stabilized format (e.g., onto TD tubes) and sent to a central lab for confirmatory analysis and precise quantification using GC-MS, which provides the definitive identification required for clinical decision-making [1] [68].
- Environmental and Indoor Air Quality Monitoring: GC-IMS is highly effective for continuous, on-site monitoring of VOCs in indoor air (e.g., in offices, schools, or industrial facilities) due to its portability and real-time capability [9]. When hazardous compounds like benzene or formaldehyde are detected at levels of concern, GC-MS provides the legally defensible, quantitative data needed for regulatory compliance and risk assessment, thanks to its broader linear range and high quantification accuracy [9] [66].
- Pharmaceutical Analysis and Food Quality Control: In pharmaceutical manufacturing, GC-MS is the established workhorse for quantifying residual solvents and mutagenic impurities to strict regulatory standards (e.g., ICH Q3C) [65]. GC-IMS finds a complementary role as a Process Analytical Technology (PAT) tool for real-time monitoring of reaction kinetics or solvent purging during synthesis, enabling rapid process adjustments [65]. Similarly, in food science, GC-IMS can rapidly profile and compare VOC fingerprints to monitor spoilage or authenticity, while GC-MS provides detailed, compound-specific analysis for in-depth quality control [69].
GC-IMS and GC-MS are not competing technologies but rather complementary pillars of a modern analytical strategy. GC-IMS serves as a highly sensitive, rapid sensor for on-site screening and high-throughput fingerprinting, while GC-MS acts as the definitive identifier and quantifier in the laboratory. By implementing them synergistically—using GC-IMS to guide targeted sampling for the more resource-intensive GC-MS—researchers and professionals can achieve unprecedented efficiency, coverage, and confidence in VOC analysis across medical, environmental, and industrial domains. This integrated approach effectively bridges the gap between the immediate need for field intelligence and the rigorous demands of laboratory confirmation.
Conclusion
GC-IMS emerges as a disruptive technology that successfully bridges the critical gap between laboratory-grade analysis and rapid on-site screening. Its unparalleled speed, high sensitivity, and portability make it ideal for initial field assessments, triage, and real-time monitoring, as demonstrated in forensic and quality control applications. However, its limitations in compound identification and susceptibility to matrix effects necessitate a complementary relationship with lab-based GC-MS for definitive confirmation and broad-spectrum untargeted analysis. The future of analytical science lies in integrated workflows, where GC-IMS acts as a rapid, sensitive front-line sensor, guiding more targeted and efficient use of central laboratory resources. For biomedical and clinical research, this promises faster diagnostic pathways, real-time monitoring of volatile biomarkers, and accelerated drug development cycles, fundamentally enhancing responsiveness and decision-making capabilities.
References
- 1. GC-IMS in Medicine: Transforming Diagnostics with ... [https://pubmed.ncbi.nlm.nih.gov/40720173/]
- 2. Recent development of HS-GC-IMS technology in rapid ... [https://www.sciencedirect.com/science/article/abs/pii/S0165993621002582]
- 3. Full Workflows for the Analysis of Gas Chromatography—Ion ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8469025/]
- 4. Gas Chromatography and Ion Mobility Spectrometry: A Perfect Match? [https://www.chromatographyonline.com/view/gas-chromatography-and-ion-mobility-spectrometry-a-perfect-match-]
- 5. - GeeksforGeeks Gas Chromatography [https://www.geeksforgeeks.org/chemistry/gas-chromatography/]
- 6. Comparison of the quantification performance of thermal desorption... [https://link.springer.com/article/10.1007/s00216-025-05933-w]
- 7. Ion mobility spectrometry [https://en.wikipedia.org/wiki/Ion_mobility_spectrometry]
- 8. Ion Mobility Spectrometry: Fundamental Concepts ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6832852/]
- 9. Gas Chromatography – Ion Mobility Spectrometry as a tool ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9647320/]
- 10. Ion mobility-mass spectrometry to extend analytical ... [https://www.sciencedirect.com/science/article/pii/S002196732200694X]
- 11. A hyper-fast gas chromatograph coupled to an ion mobility ... [https://www.sciencedirect.com/science/article/pii/S0021967324007507]
- 12. Revealing illicit drug laboratories by gas chromatography- ... [https://www.sciencedirect.com/science/article/pii/S0379073825003056]
- 13. The Combined Analysis of GC-IMS and GC-MS Reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11311445/]
- 14. Ion Mobility Spectrometers as Chromatographic Detectors [https://www.spectroscopyonline.com/view/ion-mobility-spectrometers-chromatographic-detectors]
- 15. Comparison of portable and benchtop GC–MS coupled to ... [https://www.sciencedirect.com/science/article/abs/pii/S246817092030028X]
- 16. Is GC-IMS the new Swiss army knife of gas phase analysis? [https://www.sciencedirect.com/science/article/abs/pii/S0165993623005253]
- 17. The Rise of GC–IMS as a Sustainable Alternative to GC–MS [https://www.chromatographyonline.com/view/an-inside-look-at-green-analytical-chemistry-the-rise-of-gc-ims-as-a-sustainable-alternative-to-gc-ms]
- 18. Comparison of the quantification performance of thermal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12276119/]
- 19. Towards a hand-held, fast, and sensitive gas chromatograph-ion mobility spectrometer for detecting volatile compounds | Analytical and Bioanalytical Chemistry [https://link.springer.com/article/10.1007/s00216-020-03059-9]
- 20. The Combined Analysis of GC-IMS and GC-MS Reveals ... [https://www.mdpi.com/2304-8158/13/15/2364]
- 21. Review on Ion Mobility Spectrometry. Part 1: Current Instrumentation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4331213/]
- 22. Automated 2D peak detection in gas chromatography-ion ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267024000059]
- 23. Comparison of Sensitivity Of Headspace GC, Purge and ... [https://www.sisweb.com/referenc/applnote/app-39.htm]
- 24. Dynamic Headspace vs. Purge and Trap: A Comparative Insight [https://www.gerstelus.com/dynamic-headspace-vs-purge-and-trap-a-comparative-insight/]
- 25. Comparison Sensitivity Headspace GC,P&T TD & DTE [https://www.sisweb.com/referenc/abstract/cmpsnstv.htm]
- 26. A rapid and on-site detection of pesticide residue from fruit ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X22007470]
- 27. Use of GC-IMS for detection of volatile organic compounds ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8897540/]
- 28. Recent Advances in Ion Mobility Instrumentation for ... [https://www.sciencedirect.com/science/article/abs/pii/S0165993625003759]
- 29. – GC facilitates identification of carbapenem-resistant Klebsiella... IMS [https://amb-express.springeropen.com/articles/10.1186/s13568-024-01708-1]
- 30. Determination of Methamphetamine by High-Performance Liquid... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10934924/]
- 31. Gas chromatography–trapped ion mobility mass spectrometry [https://www.sciencedirect.com/science/article/abs/pii/S0045653525005016]
- 32. Ultrafast Gas Chromatography-Tandem Differential Mobility ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11359381/]
- 33. Mapping of Urinary Volatile by a Rapid... Organic Compounds [https://pmc.ncbi.nlm.nih.gov/articles/PMC9699414/]
- 34. Analysis and application of volatile metabolic profiles of Escherichia... [https://pubs.rsc.org/en/content/articlehtml/2024/ra/d4ra03601h]
- 35. Matrix-Assisted Laser Desorption Ionization-Time of Flight ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1392959/]
- 36. Rapid on-site identification of hazardous organic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7476632/]
- 37. Rapid evaluation of mold deterioration in maize using GC- ... [https://www.sciencedirect.com/science/article/abs/pii/S2212429225019844]
- 38. GC-IMS identification of early-warning biomarkers and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12307484/]
- 39. Characterizing volatile in oral cancer... organic compound profiles [https://www.nature.com/articles/s41598-025-15510-x?error=cookies_not_supported&code=c871fa59-159a-4473-88b3-26c2ebbd97fd]
- 40. Reference library for suspect screening of environmental ... [https://www.nature.com/articles/s42004-025-01619-7]
- 41. Illicit Drug Analysis Using GC–IMS and Oral Fluids [https://www.chromatographyonline.com/view/illicit-drug-analysis-using-gc-ims-and-oral-fluids]
- 42. Mass Spectrometry Market Size, Share & Growth 2025-2030 [https://marksparksolutions.com/reports/china-mass-spectrometry-market]
- 43. Non-Targeted Screening Approaches for Profiling of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8468721/]
- 44. Ion-Mobility Spectrometry - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/ion-mobility-spectrometry]
- 45. Review on atmospheric pressure ionization sources for gas ... [https://www.sciencedirect.com/science/article/pii/S0003267022009242]
- 46. The effect of humidity on gas sensing with ion mobility ... [https://www.sciencedirect.com/science/article/abs/pii/S0925400515005547]
- 47. GCIMS: An R package for untargeted gas chromatography [https://www.sciencedirect.com/science/article/pii/S0169743923001880]
- 48. Chemometrics for ion : recent advances... mobility spectrometry data [https://pubs.rsc.org/en/content/articlehtml/2016/an/c6an01008c]
- 49. iMatch: A retention index tool for analysis of gas... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3163042/]
- 50. Gas Chromatography–Ion Mobility Spectrometry for Food Flavor Analysis [https://www.news-medical.net/life-sciences/Gas-chromatographye28093ion-mobility-spectrometry-for-Food-Flavor-Analysis-A-New-Frontier.aspx]
- 51. Pushing Peak Shapes to Perfection by High-Temperature ... [https://www.mdpi.com/2227-9040/13/4/131]
- 52. Evaluation of IMS drift tube temperature on the peak shape ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914023001480]
- 53. Go With the Flow: Thinking About Carrier Gas Flow in GC [https://www.chromatographyonline.com/view/go-flow-thinking-about-carrier-gas-flow-gc]
- 54. Chiral Trapped-Headspace GC-QMS-IMS [https://www.mdpi.com/2227-9040/12/8/165]
- 55. variable extraction strategies for dimensionality reduction ... [https://www.sciencedirect.com/science/article/abs/pii/S0963996922008377]
- 56. Coupling Front-End Separations, Ion Mobility Spectrometry ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5627998/]
- 57. Comparison of the quantification performance of thermal desorption... [https://pubmed.ncbi.nlm.nih.gov/40461898/]
- 58. Dopant-Assisted Ion Mobility Spectrometry [https://www.mdpi.com/1420-3049/29/18/4482]
- 59. What is Gas Chromatograph-Ion Mobility Spectrometer ( GC - IMS )? [https://www.linkedin.com/pulse/what-gas-chromatograph-ion-mobility-spectrometer-4mvde]
- 60. A dual-polarity ion mobility spectrometer for capturing ... [https://www.sciencedirect.com/science/article/pii/S0021967325002717]
- 61. Comparison of GC-MS and GC×GC-MS in the Analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4849136/]
- 62. Benchmarking machine learning methods for ... [https://www.sciencedirect.com/science/article/abs/pii/S002196731930158X]
- 63. Portable Electrochemical Device for Rapid and On-Site ... [https://www.sciencedirect.com/science/article/abs/pii/S0925400525014856]
- 64. Assessing the Impact of Measurement Precision on Metabolite ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12242904/]
- 65. GC-MS applications in pharmaceutical analysis [https://www.europeanpharmaceuticalreview.com/article/77425/gc-ms-applications-in-pharmaceutical-analysis/]
- 66. What Are the Advantages of Gas Chromatography-Mass ... [https://www.mtoz-biolabs.com/what-are-the-advantages-of-gas-chromatography-mass-spectrometry.html]
- 67. Why GC-MS is Ideal for Real-Time Process Gas Analysis [https://www.jeolusa.com/NEWS-EVENTS/Blog/why-gc-ms-is-ideal-for-real-time-process-gas-analysis]
- 68. A comparison between mobile and stationary gas ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9672629/]
- 69. Evaluating Ice-Temperature Storage Efficacy on Volatile Compounds in... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11988682/]